
Researchers from the University of Greifswald, Germany, introduced a new version of the BRAKER software BRAKER3, notably improving the annotation of eukaryotic genomes. Its superior performance in eukaryotic genome annotation, particularly for species with large and complex genomes, signifies a promising advancement in bioinformatics with far-reaching implications for genomic research and applications in diverse fields.
BRAKER3: A Comprehensive Annotation Pipeline
Eukaryotic gene prediction is challenging due to the complexity of large genomes and diverse data. It integrates protein homology, transcriptomes, and genomics for precise annotations. BRAKER3 annotates protein-coding genes in eukaryotic genomes by integrating short-read RNA-Seq and a large protein database coupled with iteratively learned statistical models tailored for the specific genome. Benchmarking on 11 species demonstrated BRAKER3’s superior performance over BRAKER1, BRAKER2, MAKER2, and Funannotate, particularly for species with large and complex genomes. The user-friendly Singularity container simplifies installation, establishing BRAKER3 as an accurate and accessible eukaryotic genome annotation tool.
The Genomic Annotation Gap
While eukaryotic genome sequencing soars, annotation struggles to keep pace, leaving a significant portion of eukaryotic species without annotations. This gap highlights the urgent need for highly automated and reliable annotation pipelines, especially for projects like the Earth BioGenome Project, which aims to annotate a vast number of eukaryotic species.
Current pipelines combine intrinsic and extrinsic evidence—transcripts and homologous proteins—but gaps persist. Computational predictions, though error-prone, are the backbone of traditional Ab-initio methods like AUGUSTUS and GeneMark-ES. Bridging the gap entails integrating these methods with extrinsic evidence, promising improved prediction accuracy.
Integration of Extrinsic and Intrinsic Evidence
The earlier BRAKER1 and BRAKER2 pipelines integrated either RNA-Seq or homologous proteins to enhance gene prediction accuracy. Recognizing the potential for greater accuracy through the integration of both RNA-Seq and protein evidence, a combiner tool, TSEBRA, was developed to merge predictions from BRAKER1 and BRAKER2, utilizing both types of extrinsic evidence. Subsequently, the GeneMark-ETP pipeline demonstrated significant advancements by integrating both sources of extrinsic evidence and incorporating novel approaches to training set generation and model training, particularly beneficial for species with large and complex genomes.
Motivated by these advancements and the popularity of the previous BRAKER tools, BRAKER3 was developed. It leverages GeneMark-ETP, AUGUSTUS, and TSEBRA to create a novel workflow that integrates transcript and protein homology extrinsic evidence, enhancing gene prediction accuracy. Comparative benchmarking against existing tools, including MAKER2 and Funannotate, showcased BRAKER3’s superior performance, especially for species with intricate genomes. Additionally, the provision of a Singularity container for BRAKER software streamlines installation, enhancing BRAKER3’s accessibility and cementing its position as an accurate and user-friendly tool for eukaryotic genome annotation.
BRAKER3: An Integrated Gene Prediction Pipeline
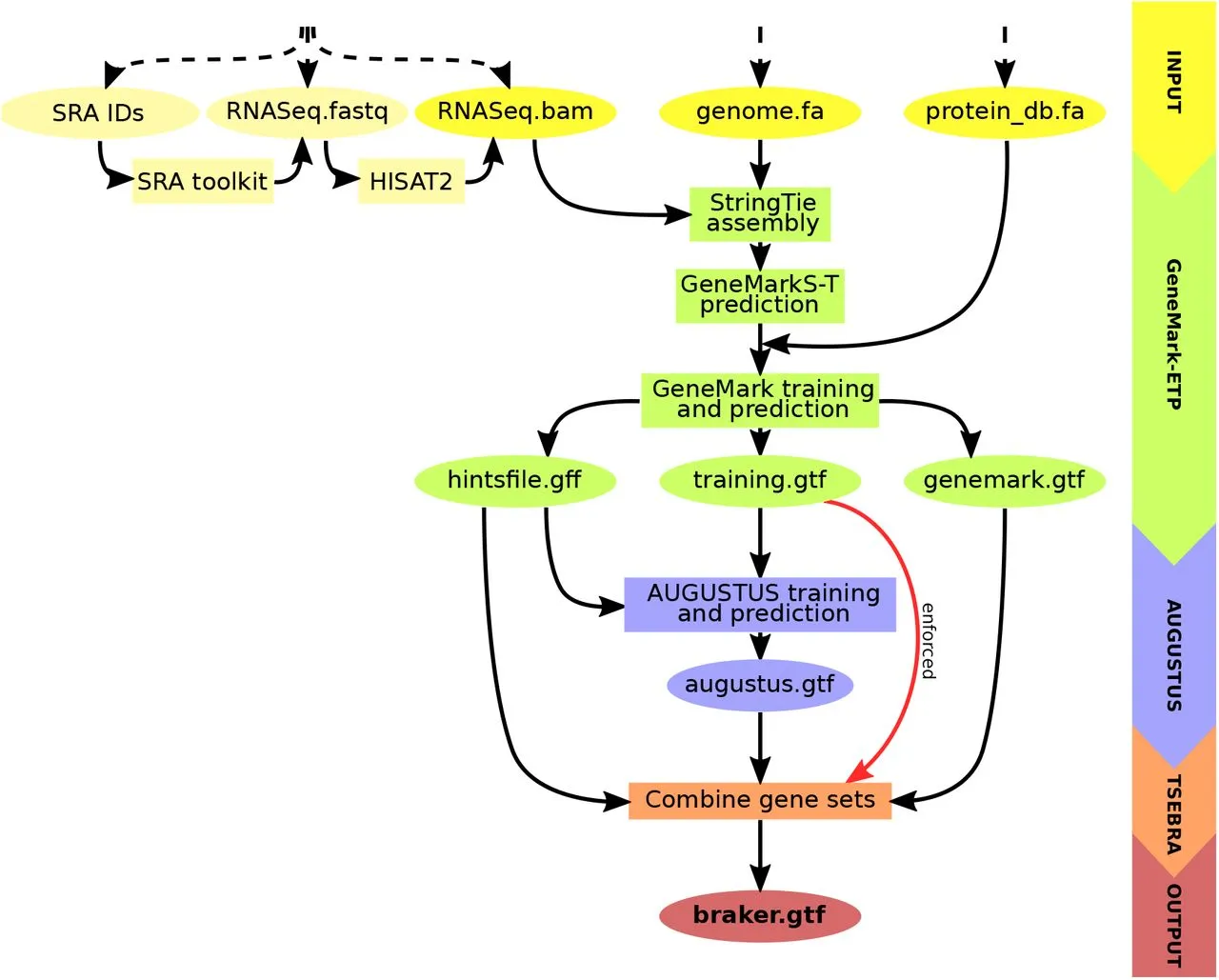
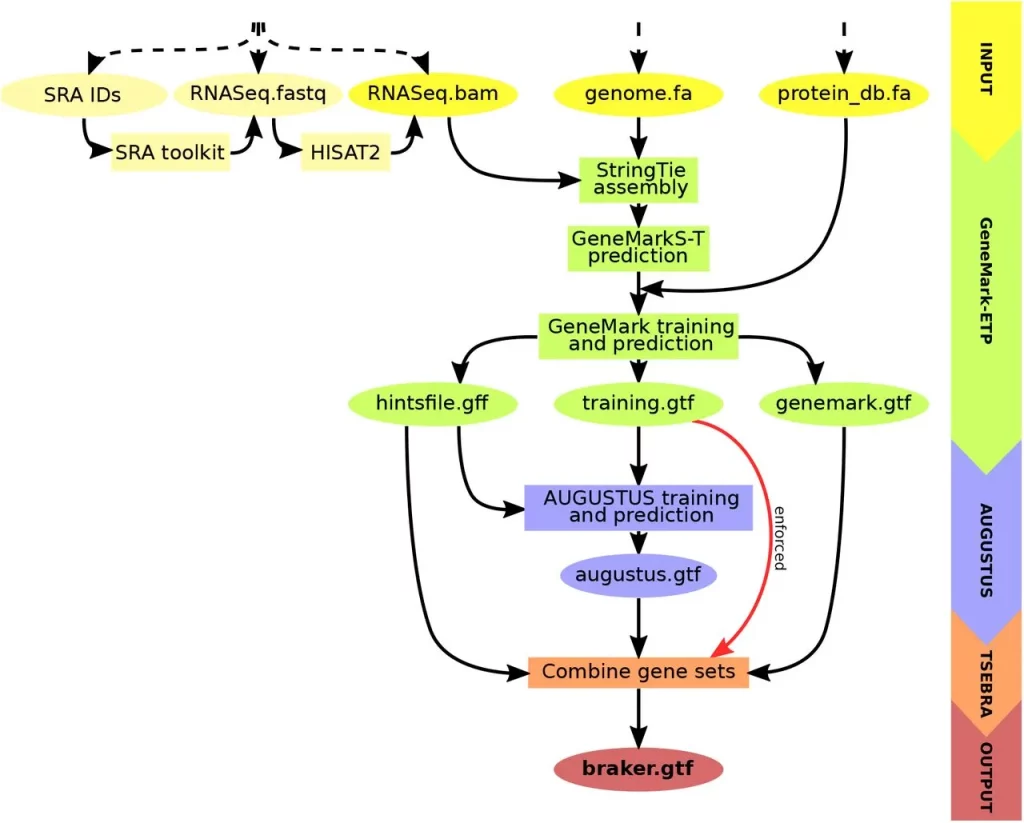
BRAKER3, the newest member of the BRAKER family, revolutionizes gene prediction. It uses GeneMark-ETP to generate various external suggestions using a target genome, short-read RNA-Seq data, and a protein database. High Confidence (HC) genes can be predicted using these hints, which come from a variety of alignments and StringTie2 transcripts. Then, AUGUSTUS is trained on this list of HC genes to produce a thorough genome-wide gene prediction. AUGUSTUS and GeneMark-ETP predictions are combined via TSEBRA in the end, ensuring that HC genes are incorporated into the outcome. BRAKER3 is a cutting-edge, comprehensive gene prediction pipeline with broad ramifications.
Key Steps in BRAKER3
- Input Data Preparation: Requires genome sequence, short-read RNA-Seq datasets, and a protein database file.
- Hint Generation with GeneMark-ETP: Utilizes spliced alignments from both RNA-Seq and database proteins to generate various types of external hints.
- High Confidence Gene Prediction: Predicts HC genes from assembled transcripts based on alignment scores and additional criteria.
- AUGUSTUS Training and Genome-Wide Prediction: AUGUSTUS is trained on HC genes and predicts a genome-wide gene set using external hints.
- Integrated Prediction with TSEBRA: An updated TSEBRA combines AUGUSTUS and GeneMark-ETP predictions while ensuring direct inclusion of HC genes for accurate genome-wide gene prediction.
TSEBRA: Enhancing Transcript Selection in BRAKER
TSEBRA, the Transcript SElector for BRAker, has been refined and expanded in its application within the BRAKER suite. It evaluates and consolidates candidate transcript isoforms by comparing transcript structures based on four transcript scores. These scores measure the alignment of transcript structures with extrinsic evidence, specifically focusing on intron position intervals. A significant improvement is the introduction of normalization for these transcript scores, considering all input gene sets. This normalization allows for better evaluation of evidence support relative to the available evidence for the target genome. Additionally, a new feature filters out single-exon genes without start- or stop-codon hint support, enhancing accuracy, especially for larger genomic sequences. TSEBRA has been integrated into the workflows of BRAKER1 and BRAKER2, combining AUGUSTUS predictions with well-supported transcripts from GeneMark-ET/EP, further enhancing gene prediction accuracy.
Test Data for Accuracy Assessment
Test data for assessing accuracy in 11 target species were compiled, including genome assemblies, randomly selected RNA-Seq libraries, protein databases, and reference genome annotations. Repeats in genomic sequences were soft-masked using RepeatModeler2 before experiments. Three protein databases were prepared for each species, encompassing sequences from the same taxonomic clade. Two large databases utilized a broad partition of OrthoDB, while the third smaller database, “close relatives included,” involved a curated selection of closely related species for efficient comparisons with BRAKER3, MAKER2, and Funannotate. Notably, challenges with running Funannotate on large protein databases were observed, prompting the creation of the third series of databases to facilitate comprehensive three-way comparisons.
Experiments and Comparative Evaluation
The performance of BRAKER3 was thoroughly evaluated and compared against seven other methods: BRAKER1, BRAKER2, TSEBRA, MAKER2, FINDER, Funannotate, and GeneMark-ETP. Three sets of experiments were conducted to compare these methods. In the first set, BRAKER3, BRAKER2, FINDER, and GeneMark-ETP were tested using large protein databases (order excluded and species excluded) to assess their accuracy. The second set compared BRAKER3 with MAKER2 and Funannotate using smaller target-specific databases, focusing on close relatives included. Additionally, variant sets of gene predictions were obtained for Funannotate by utilizing recommended flags, and the best-performing variant was considered. Accuracy metrics such as sensitivity (Sn), specificity (Sp), and F1-score were employed to evaluate the accuracy of gene predictions at the exon, gene, and transcript levels, using reference annotations for comparison. The evaluation criteria were defined to assess both individual transcripts/exons and overall gene accuracy.
Intriguing Results and Insights
Using Large Protein Databases
In evaluations with large protein databases, the order excluded database provided a conservative accuracy estimate, mirroring scenarios for novel eukaryotic genomes. BRAKER3 significantly outperformed BRAKER1 and BRAKER2, particularly in species with GC-heterogeneous or large genomes. Notably, in Gallus gallus, BRAKER3 showed a substantial accuracy boost, benefiting from GeneMark-ETP’s improvements. AUGUSTUS predictions in BRAKER3, trained with highly specific HC genes from GeneMark-ETP, surpassed GeneMark-ETP’s accuracy. TSEBRA integration into BRAKER3 further enhanced sensitivity and specificity, excelling in accuracy at both gene and transcript levels. While using the species-excluded protein database slightly improved overall accuracy, BRAKER3 remained the most accurate, underscoring its practical advantage and enhanced performance.
Using Small Protein Databases
In the scenario with smaller protein databases (close relatives included), Funannotate and MAKER2 faced challenges in completing annotations for all genomes. Comparing BRAKER3 with the successful runs of Funannotate and MAKER2 on eight species, BRAKER3 consistently outshone both in exon, gene, and transcript level accuracy. BRAKER3 exhibited significant F1-score improvements over Funannotate, emphasizing its superior accuracy. Despite the challenges, BRAKER3 showcased reliability and effectiveness, proving its robustness in eukaryotic genome annotation, particularly in various data and database settings.
Efficient Execution: Speed and Adaptability
BRAKER3 demonstrated reasonable runtimes, ranging from 5h 37min to 64h 16min, making it suitable for annotating even large genomes. Comparatively, it required only 23% more time for large protein databases than smaller ones. When benchmarked against Funannotate and MAKER2 on close relatives, including protein databases, BRAKER3 showed efficient runtime, even rivaling MAKER2 on similar hardware. A Singularity container for BRAKER is provided to address installation challenges, enhancing accessibility and ease of use in diverse computing environments.
BRAKER3: A Milestone in Genome Annotation Tools
BRAKER3 represents a significant advancement in eukaryotic genome annotation, integrating diverse data sources and leveraging innovative combiners and models to achieve remarkable accuracy. The successful benchmarking against various existing tools underscores its potential to revolutionize the annotation landscape, making it a valuable asset for the genomics community.
Article source: Reference Paper | BRAKER3 is available on GitHub and as a Docker/Singularity container.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Prachi is an enthusiastic M.Tech Biotechnology student with a strong passion for merging technology and biology. This journey has propelled her into the captivating realm of Bioinformatics. She aspires to integrate her engineering prowess with a profound interest in biotechnology, aiming to connect academic and real-world knowledge in the field of Bioinformatics.











{kind=link}