
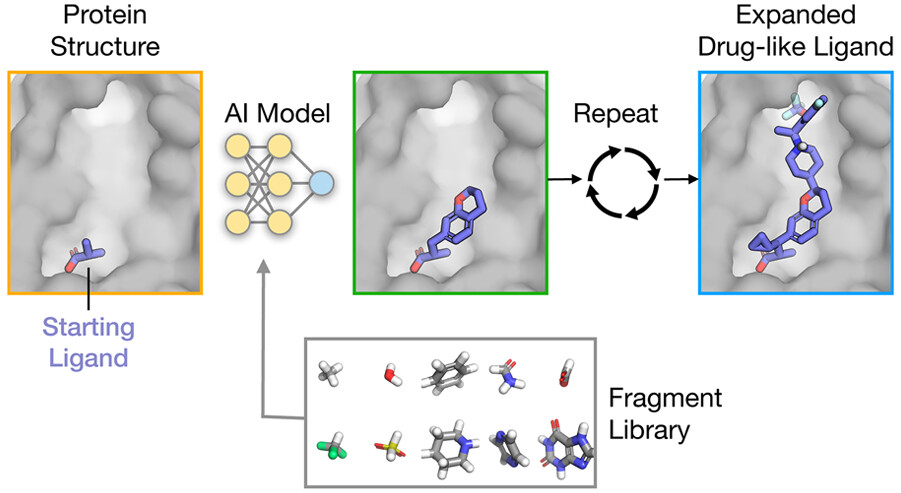
Stanford University researchers developed FRAME (Fragment-Based Molecular Expansion), a machine learning framework that uses three-dimensional structures of interactions between proteins and ligands to facilitate drug design based on ‘fragments.’ Fragments are singular chemical groups that are added to ligands for the purpose of performing lead optimization tasks and improving their properties. Ligands are small molecules that bind to different types of molecules at their respective receptors. A long-standing challenge in drug design has long been trying to answer the question of how exactly we expand ligands to study them better; FRAME will attempt to solve this and more. Using in-built Machine Learning algorithms, FRAME iteratively decides which fragments will be added to the ligand and the site at which they will be located. It simultaneously predicts the geometry of these fragments as well. The extended molecules generated by FRAME have shown improved rates of predicted selectivity and binding affinity, better than molecules generated through molecular docking, all while requiring little to no prior knowledge regarding ligand-molecule interactions.
An overview of drug discovery – and its challenges
The researchers in this field encounter numerous hurdles and long and arduous work hours during the drug discovery process. Choosing the compounds that can be employed to advance testing and medication development is a major difficulty. Considering the gigantic and ever-increasing size of chemical libraries, it is a mammoth task to screen the entire space to identify the optimal molecules to use. It still needs to be done in order to proceed with developing therapeutic treatment approaches and to save money and time.
Fragments and their role in drug design
An important step in the drug design process is the addition of small ‘bits’ of chemical groups, hereby termed ‘fragments,’ to track various properties of ligands, such as their solubility and binding affinity. Lead optimization is one such method of adding fragments, where they are added in an iterative manner based on any modifications and require further rounds of testing on the molecule. Usually, multiple fragments are added to enhance this process. However, A challenge with this method is the near-infinite number of permutations and combinations of chemical modifications that can be performed on the molecule, making it once again a difficult task to determine which modification can give the most desired result.
A look into the framework of FRAME
The expansion of the ligand is represented as a successive order of steps in the three-dimensional space. The researchers applied neural networks and trained them to understand patterns observed in the binding of high-affinity ligands to their respective proteins, which exhibit characteristics of drugs as well, therefore termed ‘drug-like’ compounds. This is a stark contrast to traditional methods that provide strict parameters for determining the affinity of a molecule. According to the experts, the more data that is fed into FRAME, the more accurate and efficient it will become. This means that there will be more data available for us to use. Another feature of FRAME that can help biologists and chemists gain a head start on their experimental operations is its ability to generate hypotheses quickly.
How FRAME is different from other approaches
The researchers highlight that FRAME is novel in comparison to other standard methods for determining the affinity of ligands, such as ligand docking, a protocol that involves the use of scoring functions inspired by physics to perform virtual screening of compounds. They argue that FRAME reduces the time spent on a single compound and does not waste time analyzing each and every one of them, as opposed to what is done in virtual screening; this gives FRAME an edge over said method as it explores the chemical space on a much larger scale in a shorter period of time. To put this into perspective, FRAME is capable of expressing over 300 billion molecules in just five simple expansion steps. Had docking been used to perform the same task, it would have taken several weeks or even a month to get it done.
The researchers also note that conventional rules crafted by experts, such as docking scores used in virtual screening, can possibly contain biases, which reduces their reliability when exploring vast chemical spaces. FRAME does not employ any of these methods and, therefore, factors in greater levels of reliability compared to the aforementioned method of screening.
The competence of FRAME
Prior to applying FRAME to add whole fragments iteratively, the researchers ran a few tests to evaluate the extent to which the algorithm can perform simple tasks. They found that it is capable of identifying attachment points within protein pockets that have not been filled yet for reference ligands with high levels of accuracy. The interactions between the protein and ligand and the subsequent steric effects can be evaluated by FRAME by studying the structure of the protein pocket, and the range in which chemicals can be synthesized can be determined through information obtained on the atoms of ligands. Both of these instances show that FRAME can perform analysis on an atomic level for ligands and proteins. It also ranks the fragments selected by it in a manner that takes key interactions and factors such as the presence of electron donors and acceptors into consideration, despite no prior knowledge being fed to it regarding this subject other than a few bits on the geometries of the chemical structures.
Conclusion
Like most algorithms under development, FRAME is not perfect and has its shortcomings. It has produced ligands of poor quality on a few occasions, which resulted in lower docking scores and reduced structural complexity. It is prone to missing out on critical interactions and blocking trajectories of growth, which further hinders the accuracy of the results produced as output by FRAME. The researchers hypothesize that these drawbacks might be addressed by combining FRAME with techniques that build structures worldwide, including denoising diffusion probabilistic models. In order to provide results with more accuracy in the future and to eventually integrate them into the drug design and development domain, the researchers also want to update the datasets that were used to train the networks within FRAME.
Article Source: Reference Paper
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.











{kind=link}
Nice