
Pangenomic annotation is critical for understanding the bacterial genomic variety and depends on recognizing core and accessory genes through gene prediction and clustering. These are vital for characterization and epidemiological examinations. Accessory genes tracked down in unambiguous subsets are related to attributes like antimicrobial opposition, harmfulness, and immunization escape. Current techniques include quality forecast and pangenome annotation as discrete cycles, requiring reliable expectations and comments across orthologous qualities. Quality forecast tools like Flash and Intemperate find coding arrangements, however, there are certain limitations to these.
Annotation tools like Prokka and DFAST integrate gene prediction with functional labeling however, they need consistency in expectations and explanations across orthologs. Mistakes in expectation and explanation block bunching and useful derivation. Due to this, further developing clustering quality is crucial for improving pangenomic annotation, working with transformative models, and anticipating reactions like vaccines in bacterial populations.
Pangenome analysis has primarily depended on gene prediction inside individual genomes, prompting overt repetitiveness and failure. Pangenome graphs like De Bruijn charts (DBGs) have been presented to address this, considering a more effective and nonredundant portrayal of population variety. ggCaller is a novel tool that uses DBGs, which work on populace recurrence data for further improved gene prediction. It recognizes homologous beginning codons and works with steady scoring and practical explanation of orthologs. ggCaller also offers a query mode for reference-agnostic functional inference, making it valuable for pangenome-wide association studies (PGWAS). Its precision and computational benefits are exhibited through correlations with the present status of tools utilizing simulated and genuine bacterial genomes.
Overview of the ggCaller workflow
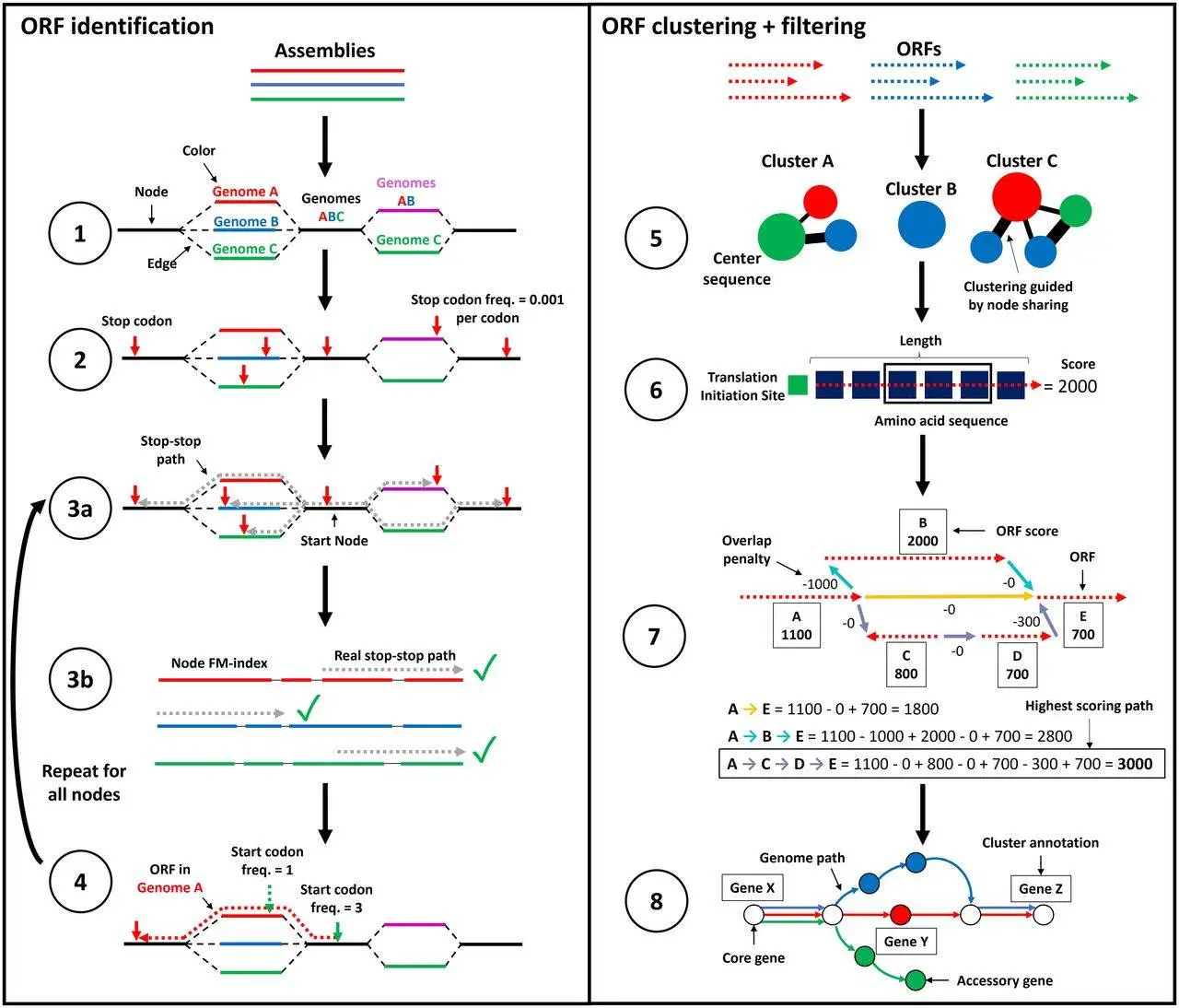
ggCaller is a gene-prediction tool that operates within a De Bruijn graph (DBG), optimizing gene prediction, clustering, and annotation of orthologous genes. In step one, DBG is constructed using Bifrost, generating nodes based on matching k-mers from input genome assemblies. Each node is “colored” to represent the genomes in which the corresponding k-mers are found, aiding in population frequency calculations.
ggCaller then identifies stop codons within the DBG and traverses the graph to pinpoint putative gene sequences or open reading frames (ORFs). A depth-first search (DFS) is employed to pair stop codons and delineate the coordinates of all possible reading frames. After this, candidate paths are cross-validated against the input sequences.
Focusing on “center sequences” or longest ORFs sharing a common node, ORFs are then clustered based on a modified Linclust approach. Edlib calculates pairwise edit distances, and ORFs cluster with the center sequence exhibiting the highest identity. Following clustering, a scoring model in Balrog generates per-residue scores for the center sequences. These scores are extended to other ORFs within the respective clusters, minimizing the need for extensive model queries.
Next, ggCaller determines the highest-scoring tiling path through the DBG per input genome, penalizing large overlaps between adjacent ORFs. This process yields a population-wide set of coding sequence (CDS) predictions. The predictions are further processed using an adapted version of Panaroo’s gene graph algorithm, directly compatible with DBGs. Clusters are refined based on a specified identity threshold, resolving paralogous clusters and eliminating poorly supported ones. Functional annotation using DIAMOND and/or HMMER3 is applied to cluster center sequences.
Notably, ggCaller innovates in multiple aspects. This approach significantly enhances gene prediction and annotation accuracy while reducing computational time. The default output includes a gene presence/absence matrix, annotated gene clusters, their sequences, and their respective locations in input linear sequences. Additionally, ggCaller allows for pangenome alignments, phylogenies, and single-nucleotide polymorphism calls to be automatically generated.
ggCaller accurately predicts genes in incompletely assembled structurally diverse operons. The accuracy of gene prediction in diverse, structurally complex operons was assessed using ggCaller, comparing it with other gene prediction tools.
Evaluation of outcomes from ggCaller
ggCaller underwent a thorough performance evaluation, revealing unique strengths that differentiate it from other tools. While it performed similarly to other tools in identifying genes within the original cps operons, it had slightly lower precision, mainly due to challenges in pinpointing gene start positions and predicting short open reading frames (ORFs).
What truly made ggCaller shine was its remarkable proficiency in dealing with fragmented capsular polysaccharide biosynthetic operons (cps). It excelled in recalling genes with precise start and end coordinates, even in cases of assembly fragmentation, which tends to challenge many gene prediction tools. This adaptability sets ggCaller apart.
In terms of predicted gene lengths, ggCaller consistently outperformed other tools by covering a larger proportion of the actual gene lengths, especially in highly fragmented assemblies. This advantage was facilitated by its utilization of Bifrost de Bruijn graphs (DBGs), enabling it to bridge gaps between contigs and establish connections across k-mers in various assemblies.
In simulated bacterial populations, ggCaller displayed exceptional performance when estimating essential genes shared by most bacteria, even when grappling with unorganized or incomplete genetic data. Its accuracy in identifying these vital genes was truly remarkable.
Furthermore, ggCaller’s accuracy was pitted against other methods in annotating intricate and repetitive genes in bacteria, including those with diverse structures. ggCaller demonstrated an impressive level of accuracy in anticipating the initiation and termination points of genes, assessing sequence congruence, and quantifying the number of sequences within each gene cluster (COG). It closely paralleled manual predictions, particularly in the instances of Pbp1a, Pbp2b, and PsrP genes. Additionally, when it came to predicting PsrP genes, ggCaller outperformed other tools, excelling in precision and the identification of specific isolates. These outcomes underscore ggCaller’s capability in annotating genes within intricate and structurally complex genomic domains.
ggCaller improves functional interpretation in pangenome-wide association studies
ggCaller also came through in a big way by helping us better understand the functional genes during pangenome-wide association studies (PGWAS), particularly when digging into antibiotic resistance in Streptococcus pneumoniae. The traditional methods lean heavily on specific references, but they can fall short when genes have different structures and locations.
Using PGWAS to explore tetracycline and macrolide resistance, where the resistance genes are scattered throughout the genome, ggCaller stepped up. It did a fantastic job of accurately figuring out important gene sequences without being tied down to just one reference point. Relying solely on a single reference often led to misleading results, which highlighted the limitations of the old way of doing things. In contrast, ggCaller’s annotation directly pointed towards the genes that were causing antibiotic resistance. This was a game-changer for PGWAS, making the interpretations more precise, regardless of which reference genome is used. For example, ggCaller correctly identified tetM for tetracycline resistance and provided accurate annotations for mefE and mel genes involved in erythromycin resistance.
The study also taught us that ggCaller’s performance is quite adaptable to the diversity of the bacterial population, which essentially depends on how intricate the de Bruijn graph (DBG) is. Afterward, ggCaller was compared to Prokka + Panaroo using different datasets that represented various levels of pangenome diversity in Streptococcus pneumoniae and Neisseria gonorrhoeae. Interestingly, ggCaller consistently turned out to be quicker in terms of runtime for all datasets, and this speed boost was even more significant when dealing with datasets that had a lot of pangenome diversity.
Conclusion
ggCaller marks a significant advancement in bacterial gene annotation and pangenome analysis. It does things a bit differently by using population-frequency information within a de Bruijn graph (DBG), as opposed to the old tools that primarily focus on individual genome analysis. It further improves accuracy, consistency, and speed while addressing issues related to redundancy and prediction. ggCaller can even provide functional annotation independently of specific references, which is a major plus point for pangenome-wide association studies (PGWAS). It also goes beyond currently existing tools, streamlines workflow, and enables more accurate interpretation of key findings. However, it is important to note that ggCaller has its limitations, especially in terms of memory usage and lack of iteration analysis. Overall, ggCaller represents a promising shift towards a graph-based approach to bacterial genomic analysis, increasing both efficiency and accuracy.
Article source: Reference Paper | Software availability: GitHub
Learn More:




Prachi is an enthusiastic M.Tech Biotechnology student with a strong passion for merging technology and biology. This journey has propelled her into the captivating realm of Bioinformatics. She aspires to integrate her engineering prowess with a profound interest in biotechnology, aiming to connect academic and real-world knowledge in the field of Bioinformatics.











{kind=link}