
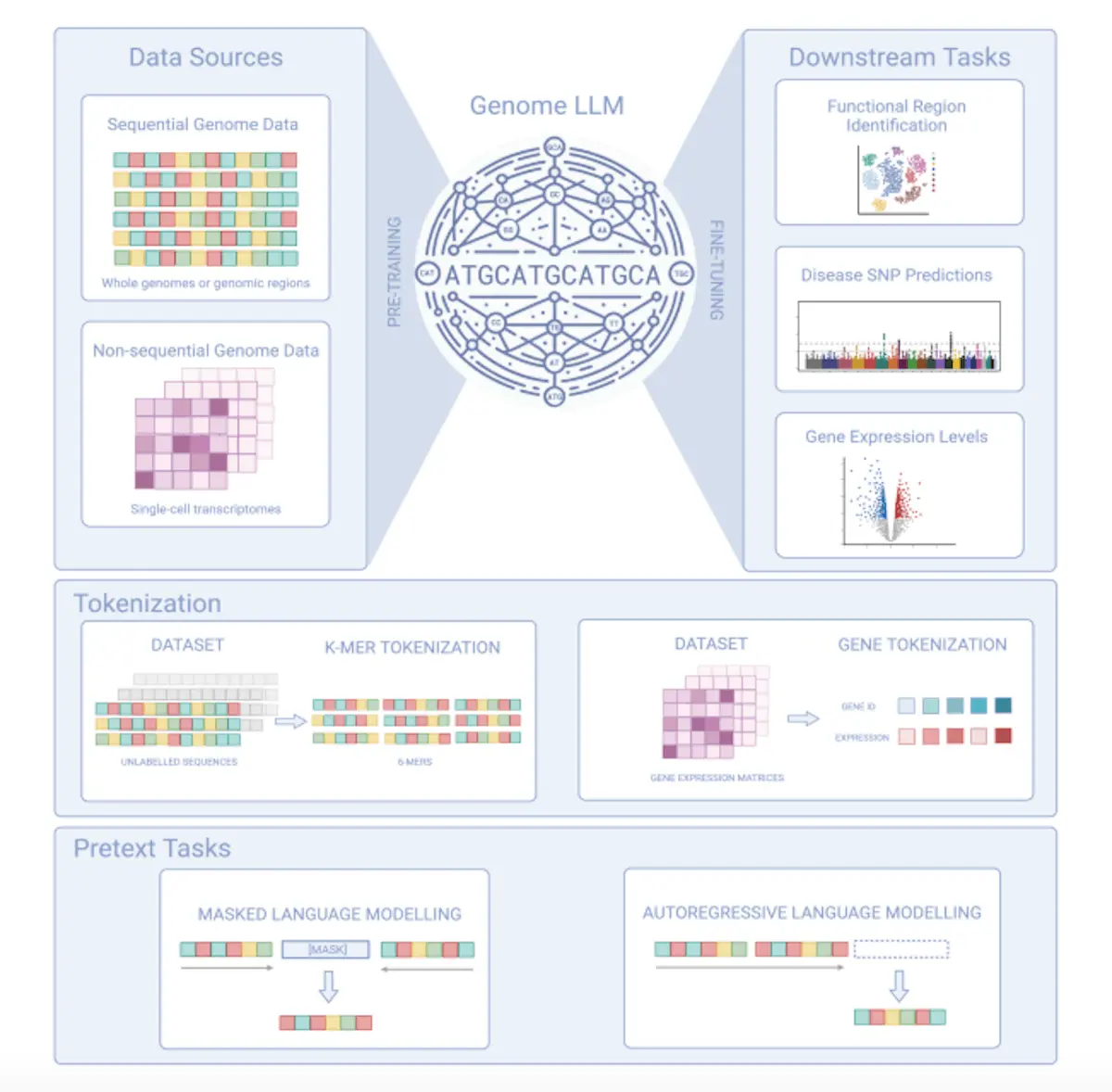
Deep learning techniques have had a significant influence on genomics and the sequencing techniques employed in the field due to the exponential advancements in artificial intelligence (AI) and machine learning (ML) during the past ten years. The review article by a team led by researchers from the University of Toronto elucidates the advantages and drawbacks of deep learning techniques utilizing both conventional convolutional neural networks and recurrent neural networks in transformers. They also discuss possible methods for genomic modeling that extend beyond transformer-based deep learning methods and hope that their paper can act as a useful tool for scientists who want to gain more insights into the applications of LLMs in genomics.
AI in everyday life
Deep-learning methods have been used in more domains than you would think; they progressed from being a niche model used and experimented on by a few individuals and programmers in computer science to being directed to perform tasks such as generating art using AI, representing different languages, and in biology, using the sequences of amino acids to predict the entire structures of proteins! One major factor that has been believed to be the driving force behind how AI tools are now a part of our everyday life for performing tasks is due to the ability of the datasets to expand rapidly based on the amount of data fed to them. This has increased the accessibility of these tools to a greater audience and has facilitated the development of multiple modes of these tools based on the function they are required to perform.
Deep-learning in genomics
Keeping in mind the infinite number of ways that AI tools can be utilized and the vast potential they hold, developers have pushed towards developing models that are bigger than before. As mentioned before, the more data you feed AI, the more reliable and useful it becomes. With more and more of these large datasets coming up, data mining is a practice scientists and researchers have warranted. In genomics, information related to the methylation of compounds, the structure of chromatin fibers, their accessibility, and the number of molecules associated with the genome acted as a source for performing data mining to study the genome better. Deep learning has emerged as a preferred technique for genomics researchers, partly because of the growing volume of data in this area and the myriad ways it can be applied to address issues with processing and sifting massive amounts of computer data.
An example of how deep learning is applied in genomics is the prediction of high-dimensional modalities when data-related DNA, RNA, or single-cell RNA sequences are entered into deep learning models. Some of the predictions made by these models include annotations with regulatory functions such as the extent of RNA and transcription-factor binding, identification of regions with promoters, and the coverage of RNA sequencing (RNA-seq) data when using information extracted from DNA sequences. Convolutional neural networks (CNNs) have commonly been used to evaluate data related to DNA. The RNA-seq data of single cells can perform a wider range of tasks, such as the annotation of cell types, predictions on the dosage sensitivity of genes, and the correction of batch effects, a lot of which can be done using different types of autoencoders.
Transformers: A revolutionary addition to the deep-learning space
Transformer-based models have been more and more popular lately because of their achievements in computer vision and natural language processing (NLP). Their application has naturally extended to genomics as well, where it can be used to perform tasks that require the production of sequences as output and also those that need to classify data and produce quantitative assays. A significant feature that makes transformers stand out over other models is their attention mechanisms, novel frameworks that involve conceptual learning in an iterative manner. This facilitates their learning of how the genome is organized thoroughly.
There are two ways in which transformers can be applied to genomic modeling: either as a standalone model or as a model present after a few layers are initially placed. Transformers can take up the former way of operation when no extension of any given context is needed when performing downstream tasks. For the latter, the initial ‘layers’ that come before the transformer layer are mainly involved in compressing all the contextual data into an embedding space of low dimension; this is required considering the computational costs that would come if big data of this sort were dealt with directly without compression. In standalone transformer models, a small number of marked ‘tokens’ are identified from the input data and processed. These models are largely involved in the development of large language models (LLMs) for genomic modeling.
Transformers can be further divided into two more classes: hybrid models and LLMs. Hybrid models consist of a complex architectural framework, of which the transformer occupies a very miniscule portion. They are designed to perform more specific tasks, such as ChIP-seq Cap Analysis Gene Expression (CAGE), and are more focused on achieving higher levels of accuracy in their results, as opposed to tasks that require more comprehensive understanding. Due to these reasons, hybrid models are not considered true LLMs.
One of the first LLMs used for analyzing the genome is DNABERT, based on the pre-existing BERT model. It has been modified to perform functions specific to DNA sequence analysis and genome modeling. These models undergo pre-training on specific information that gives context to the task that needs to be performed. This helps in accounting for dependencies in the dataset that are long-range as well as local. DNABERT has proven to be a state-of-the-art model when predicting promoter regions and transcription-factor binding sites.
Conclusion
Transformer-based LLMs have been notable due to the fact that they need to be pre-trained, which in turn has significantly contributed to understanding the composition of the genome and the functions served by certain regions much better than ever before. By highlighting conserved areas of the genome, they have also advanced evolutionary research. Steadily increasing the speed at which we comprehend the genome at deeper levels will eventually enable groundbreaking research in synthetic biology, personalized and precision medicine, and drug development.
Article Source: Reference Paper
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.











.){kind=link}