
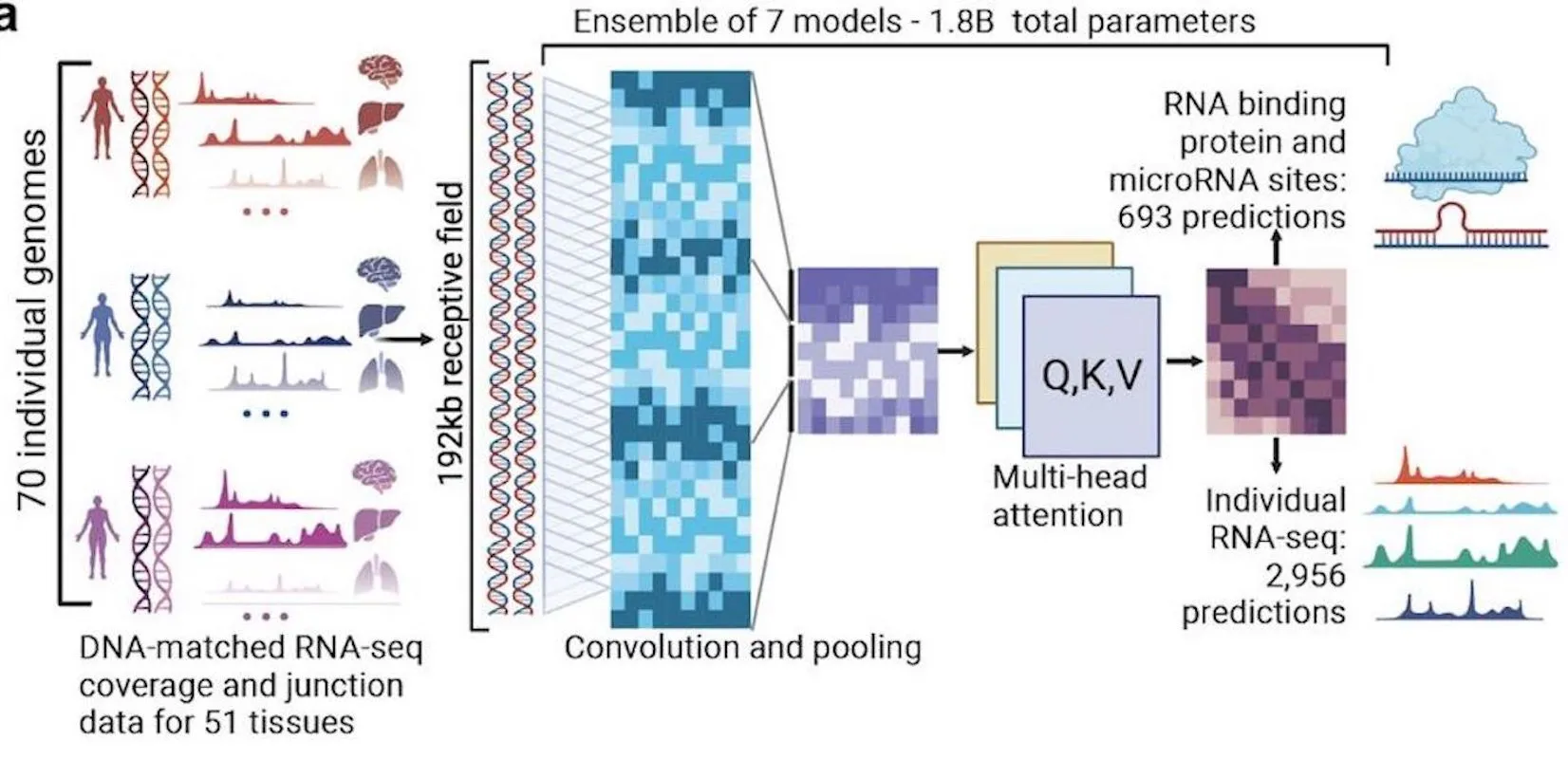
A team of researchers at Deep Genomics Inc. developed BigRNA, a foundation model for understanding the biological functions and mechanisms of RNA molecules. Two challenges faced by researchers over the years have been modeling RNA correctly and predicting its features, especially when considering interpreting genetic variations and attempting to formulate personalized therapeutics. By feeding information on DNA sequences to the model beforehand, it has been trained on multiple datasets for the purpose of predicting the expression of RNA for specific tissues, the specificity of RNA binding properties, and binding sites for microRNAs (miRNAs).
Researchers have been working on machine learning (ML) models that can use DNA sequences to predict gene expression for many years. This is now made possible with rapid advancements in artificial intelligence (AI) and deep learning (DL). They have contributed to drug discovery by giving us a much better understanding of the effects of pathogenic variants of genes on gene processing and expression and have enabled research on personalized medicine that can deal with the consequences of such variants.
Previous models lacked the ability to predict specific gene expression levels and could only predict them on a general scale. They could not detect changes that took place in regulatory mechanisms, such as transcriptional deviations observed in the processes of splicing and polyadenylation.
The benefits of RNA-sequencing methods
Data retrieved from RNA-seq methods, such as mRNA sequencing, small, total, and target RNA sequencing, have the ability to identify regulatory events in transcription that occur across a variety of genotypes. It is a resource that is easily accessible for obtaining information on RNA gene expression at a higher resolution.
An example of an extensive resource of this kind is Genotype-Tissue Expression (GTEx). It pairs RNA seq data with Whole Genome Sequencing (WGS) data. It has been difficult to build deep neural networks capable of learning RNA-Seq data to understand changes in transcriptional phenotypes.
Introducing BigRNA
It has achieved the difficult goal of understanding RNA-seq data for predicting various transcriptional events. The data is used as a training resource for the model to equip it to detect diverse regions of non-coding RNA that are pathogenic and their effects on transcription. It has applications in downstream tasks such as predicting mRNA binding sites and RNA-binding proteins (RBPs)
It is capable of directly modeling RNA-seq data, and its performance is comparable to that of specialized models used for analyzing pathogenic variants. It uses transformer-based architecture to facilitate deep learning.
Application in personalized therapeutics
Steric blocking oligonucleotides (SBOs) are a prime example of RNA-based therapeutics. The first step followed by BigRNA is the detection of compounds that induce a change in certain target regions of the genome during splicing. The model then retrieves appropriate SBO therapies that are specific to the causative compound. This is possible due to its understanding of various regulatory mechanisms. In the final step, the model designs SBOs that block regions that promote inhibition of gene expression. BigRNA does not require any training on previous cases of SBO therapy attempts.
Comparison with previous models
The same drug discovery tasks performed on BigRNA were applied to different specialized deep-learning models such as TargetScan, DeepRiPe, and SpliceAI. It had comparable performance to DeepRiPe and TargetScan and proved to be superior to SpliceAI with greater accuracy in designing SBOs and variants that skip over exons.
It can also perform the prediction of pathogenic variants in untranslated regions of genomes. It understands that the actions of these variants are indicated by a change in the polyadenylation site, which in turn reduces the variants’ half-life. It can also identify intron retention, a correlated splicing event, unlike other models.
In comparison to fine-mapping methods, it can predict any contributions made to the heritability of complex traits by analyzing different types of mechanisms. These traits may not be hinted at through structural changes in proteins.
Conclusion
There is scope to expand BigRNA applications by fine-tuning them using SBO-treatment data as a training resource. It has other potential uses in the prediction of phenotypic expression of induced ADAR (adenosine deaminase that acts on RNA) editing and mRNA designs that can increase the efficiency of translation and half-life duration.
It can also be improved by increasing the speed of transformer architecture by applying parameterized upsampling and training it on more individuals to increase its predictability across diverse sets of genotypes. Its widespread application can transform the field of drug discovery and therapeutics.
Article source: Reference Paper
Important Note: bioRiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.











{kind=link}