
A recent study published in Nature Communications explored the relationship between gene expression prediction and the use of deep learning models, which are the most advanced models or architectures in the domain of machine learning. Fritz Forbang Peleke and Jędrzej Szymański head the group to uncover the role of non-coding regulatory elements in gene expression in model plants, including Arabidopsis thaliana, plants of the nightshade family (Solanaceae), cereals (sorghum and maize) as well as, Melissa Woolley is leading an international consortium that uses modeling and data integration.
Introduction
Gene expression regulation is a complex process that comprises DNA-protein and nucleic acid-nucleic acid interactions. The processes of CREs and TFs are dedicated to transcription, and RNA processing enzymes are capable of affecting gene expression at a post-transcriptional level. The deep learning method helps to understand the mechanisms underlying this phenomenon, especially elucidating CRE annotation. Although the current trend is encouraging, large-scale discovery and CRE annotations across plants still seem to be an understudied topic. This project’s application of convolutional neural networks will be very useful for gene regulation and metabolic pathway identification of tomato(es).
Approaches Taken
Data collection involved not only genomic and transcriptomic data, which were downloaded from Ensembl Plants but also age-related transcriptome data from NCBI SRA. Models were built and trained by applying the concept of convolutional neural networks (CNNs), as well as dropout layers for regularizing the network. The main goal of the model building was to calculate the significance scores and describe the EPMs. The flanking genes were boasted by looking at the EPM state, and therefore, annotation of structural variations was carried out. Functional assessment and metabolic pathway mapping were used to this end in elucidating gene features and metabolic pathways. The statistical data in R provided support for the gene regulation processes, and it further enlightened us on how specific processes may be disrupted.
Research Insights
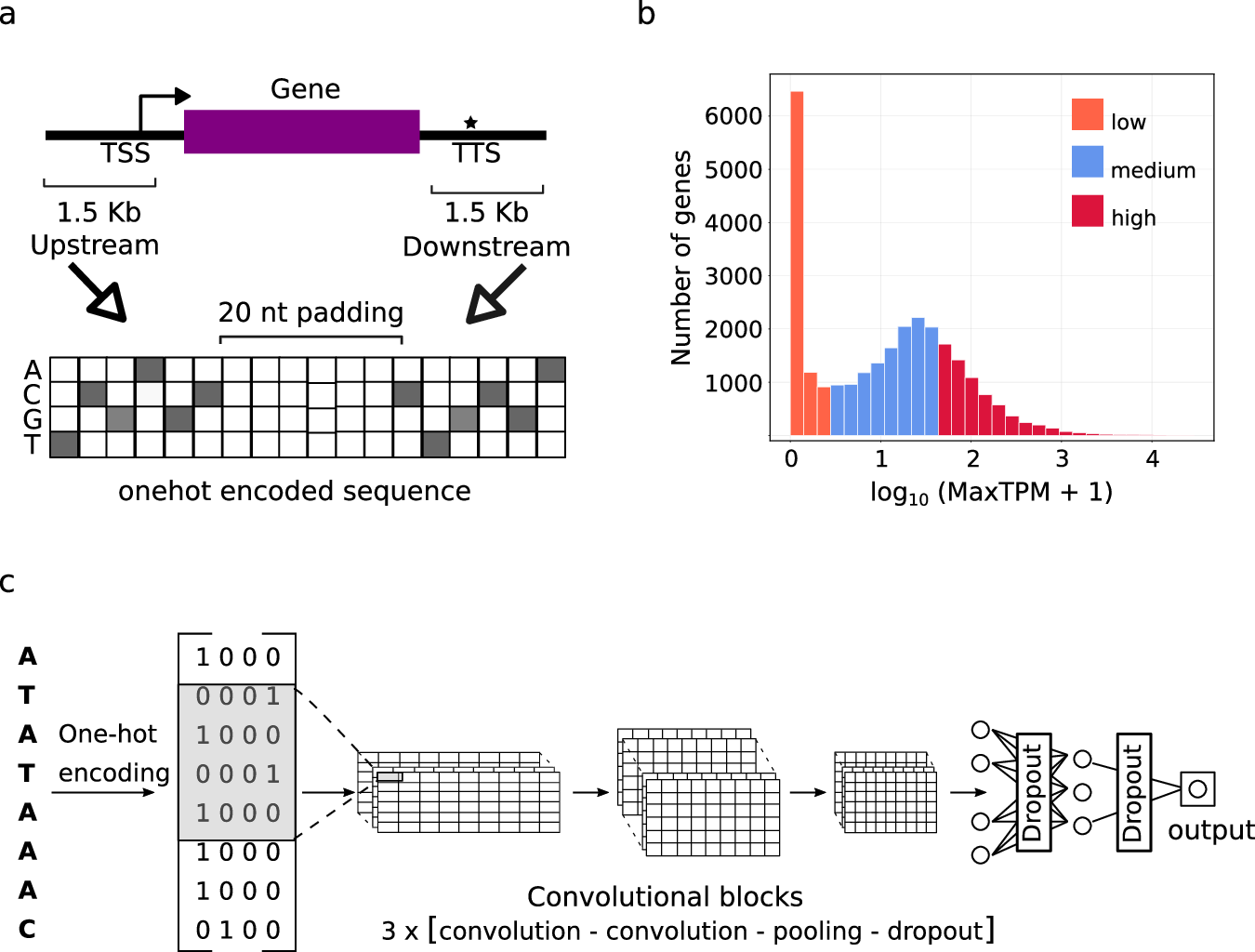
The scientists’ achievement was designing a computer model that predetermines gene expression levels in plants with nearby gene sequences. They used the genome data from Arabidopsis thaliana, Solanum lycopersicum, Sorghum bicolor, and Zea mays as training samples for their model. They got numerical sequences from both upstream and downstream regions of the beginning of transcription start and termination sites (TSS and TTS) and put them in the DNA format required for the learning of a CNN model.
The CNN model had three convolutional blocks, and each block was built with the combination of two convolutional layers acting before pooling and dropout layers. Thus, they were able to model the attributes at multiple lengths and with complexity. So, convolutional layers processed the signal, followed by a fully connected layer to predict the expression.
They then assumed two classes for the gene expression–low and highly expressed by setting the lower and upper quartiles of the gene expression distribution as 0 and 1, respectively. Cross-validation was among the training techniques employed during the process. This ensured that the model was not only robust but also generalizable so that it could be applied to more data sets other than the original training set.
The model satisfactorily predicted the expression of genes in leaves and roots across the four plant species under investigation. They also utilized forward-leaning practices to find sequence patterns that are related to expression levels. They were shown to share sequence characteristics of previously known positioned specific expression-predictive motifs known as EPMs, which were conserved across species.
The researchers also tested whether the model could be applied to predict gene expressions in species that were not in the training data by showing that different gene sequences from the same species and the ones that were similar contributed to the expression.
Additionally, they demonstrated the model’s ability to investigate the gene expression patterns of the two most closely related species, heterozygous S. lycopersicum and homozygous S. pennellii. Using the algorithm, they discovered that variation of gene expressions was dependent across various species, implying the system’s ability to get biological insights.
Through this work, we can then appraise the usefulness of simulations in forecasting and explaining gene expression cooperation in plants and thus unravel the molecular layer that conceals their existence.
Conclusion
Ultimately, this research is a considerable achievement towards unveiling the ‘language code’ required for plant gene expression in a model system. The bringing together of deep learning algorithms and genomic data discovery prospects is not only the exploration of gene regulation networks and the covering of the secrets of genetic variation.
Article Source: Reference Paper | Reference Article | Custom code used in this study is available on GitHub.
Follow Us!
Learn More:




Anshika is a consulting scientific writing intern at CBIRT with a strong passion for drug discovery and design. Currently pursuing a BTech in Biotechnology, she endeavors to unite her proficiency in technology with her biological aspirations. Anshika is deeply interested in structural bioinformatics and computational biology. She is committed to simplifying complex scientific concepts, ensuring they are understandable to a wide range of audiences through her writing.











{kind=link}