

The field of bioinformatics has seen numerous exciting breakthroughs in the year 2022 that have transformed the landscape of biological research. Many important discoveries have advanced our understanding of complex biological processes and have led to new treatments and therapies for diseases. In this article, we will explore the top 10 bioinformatics discoveries of the year and how they have impacted the field of biology. So sit back and get ready to learn about some of the most notable developments in bioinformatics!
#1. THE REMARKABLE RETELLING OF THE HUMAN GENOME: A MODERN-DAY ODYSSEY

When the human genome project officially concluded in 2003, the genome was only 92% scaled, which is a remarkable feat by itself in the then-present technological landscape. But today, as the field has advanced to incorporate those shortcomings, a consortium led by researchers at the NIH, USA, called Telomere to Telomere (T2T), was tasked to tackle the remaining 8%. The study was accomplished through the application of long-read sequencing technologies in place of short-read sequencing, where the sample DNA had to be fragmented into short fragments having a maximum of around 400 bps. The human genome story was retold with long-read sequencing approaches PacBio HiFi and Oxford Nanopore that can sequence fragments of length 20,000 and 1 million, respectively, to fill in the remaining gaps. The newly sequenced genome, termed T2T-CHM13 contained around 200 million bps, with an estimate of around 115 genes. But the story is yet to attain it’s happily ever after. The Y chromosome has again evaded being sequenced due to its absence in the study sample.
#2. ULTRA-FAST DNA SEQUENCING

In 5 hrs and 2 mins, the entire patient genome was sequenced. In 7hrs and 8 mins, the entire sequencing plus diagnosis was achieved, attributing the failing heart of the patient to myocarditis. This DNA sequencing approach, achieved at Stanford University by stringing together long-read sequencing technology, Oxford Nanopore, with a DNA analyzer that performs with 48 flow cells in tandem, is the fastest to date and has bagged the Guinness World Record for the same. The team led by Euan Ashly, who made possible this remarkable feat, plans to offer the services of sub-10 hr sequencing turnaround initially to the hospitals at Stanford and subsequently to others to improve diagnosis quality and time and are deliberation to bring down the sequencing time further.
Present like a Pro! ExpertSlides – The secret weapon for your presentations!
#3. ESMFold: META’S PROTEIN STRUCTURE PREDICTION MODEL
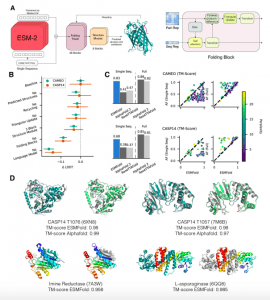
The long-accepted concept of amino acid sequence shaping the protein structure, which dictates its activity, can be employed in determining the functional impacts of mutations and in designing new drugs. But predicting the protein structure from their sequences is a daunting task that is haunted by endless possible conformations that a given protein sequence can fold into. Despite this, many teams have picked up the gauntlet with Deepmind’s AlphaFold2 reigning as the king, bringing astounding accuracy in protein structure prediction. The newest entry into the field, Meta’s ESMFold, brings tough competition by employing a language processing model, transformer, in deriving the atomic-level structure of the proteins along with confidence values from protein sequence as input. The use of protein sequences as compared to multiple sequence alignment, as in AlphaFold2, greatly reduces the computational power of ESMFold while its relatively simpler algorithm produces faster results, though AlphaFold2 produces slightly better accuracy. The availability of a number of prediction models with differing underlying algorithms makes it possible to compare and contrast to generate accurate results.
#4. AlphaFoldDB: PREDICTED STRUCTURE ATLAS OF THE PROTEIN UNIVERSE
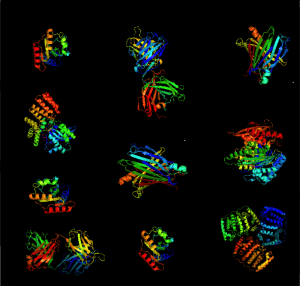
AlphafoldDB is a data repository developed as a means to make available the protein structure predictions from AlphaFold for ease of accessibility. In July of 2021, when it was initiated, it held more than 350,000 protein structure predictions, including the entire human and mouse genome. Now, over a year after its genesis, the database contains over 200 million structures accounting for 1 million species, nearly all known proteins on the planet. The 23-terabyte of contents held within the database, with a promise to accommodate more with the discovery of new organisms, is expected to produce leaps and bounds in the field of life sciences.
#5. SIMULATION OF A LIVING CELL

The prayers for a computerized model of a cell, the fundamental unit of life, to predict its biology and uncover new ones were answered through a study conducted predominantly by scientists at the University of Illinois. Understanding the importance of starting small before going big, the scientists developed a minimal cell, JCVI-syn3A, that contained only 493 genes that were termed essential for survival which was about half as that of its parental genome. The simulated model was developed through an extensive study of the cell’s protein machinery, cell wall composition, and biochemistry, all pieced together with the help of their collaborators. The developed digital simulations mimicked many of the cell behavior in an experimental setting and clashed with a few other experimental observations, all while simultaneously introducing the existence of new mechanisms and interactions. The study is aimed to advance further by incorporating not just biochemical interactions but also those interactions driven by biophysics to get a more comprehensive view of a living cell.
#6. CROPSR: A BREAKTHROUGH TOOL FOR ACCELERATED GENOME EDITING

Crispr/Cas9, the breakthrough discovery of 2020, though it holds immense potential in developing genomic variants, was predominantly focused on mammalian cells, and its use and potential in the plant kingdom are still in their infancy. In mammalian cells, where the presence of more than one copy of a gene is rare, the guide RNA sequences are penalized for having hits at more than one position within the genome. The polyploid nature of plant genomes that harbors repetitive genes and multiple chromosomes and backup genes that nullify the effect of knockdowns makes gene editing in plant systems highly laborious. Scientists from the Center for Advanced Bioenergy and Bioproducts Innovation developed the first ever open-source software, CROPSR, that can predict and evaluate guide RNA sequences from the organism’s genome. Though it was developed for targeting plant genomes, the software has also shown effective results with other genomes. The process is both computationally expensive and time-consuming but makes up for rifling through the endless online resources for a target gene, designing guides for the different locations, and performing multiple experiments. Now, the scientists could just go into their database developed from their choice of genome, search for the gene of interest, and identify all the possible guides.
#7. TRACING OUR ANCESTRY: GENOMICS-POWERED GENEALOGY BUILDS THE LARGEST FAMILY TREE
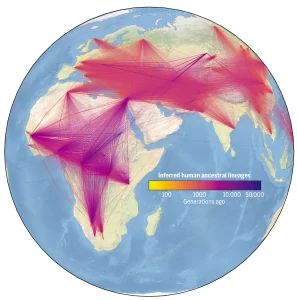
The large reservoirs of genetic data that are getting continuously replenished, if combined together, can reveal exciting discoveries held within them if and when we can surpass the hurdle of effectively merging and processing them. This feat was achieved by the scientists at Oxford’s Big Data Institute, where they developed algorithms that can be scaled to accommodate millions of such sequences. This algorithm builds the largest ever family tree to date by tracing the ancestry of genomic regions from 3,069 individual genomes that included both modern and ancestral sequences covering 215 populations to deduce patterns of variations within these regions. The study identified 27 million ancestors, while the location data of the genomes aided in capturing many notable events in human evolution. The comprehensive dataset is capable of storing millions more genomes through which scientists are aiming to map the descent of human genetic variations as seen today.
#8. AnVIL: REVOLUTIONARY BIOINFORMATICS PLATFORM PROVIDES DATA ACCESS AND ANALYSIS TOOLS TO ALL
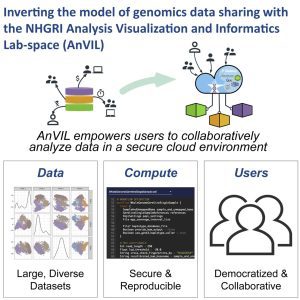
Despite its power to revolutionize the field of biological sciences, a huge roadblock in the path of Genomics is the large quantities of data it presents and the computational power and resources required to dissect and explore it. In an effort to level the playing ground and provide even small data centers equal opportunity in scientific discovery, the researchers at John Hopkins University developed a cloud-based platform, AnVIL (Genomic Data Science Analysis, Visualization, and Informatics Lab-space). On top of being one of the largest genomic databases with sequences from over 300,000 genomes, Anvil also provides resources for data analysis. It integrates tools such as Galaxy, R, Jupyter notebooks, WDLs, Gen3, and Dockstore, which allows the researchers to perform interactive genomic analysis without having to download thousands of gigabytes of data. It also promotes collaborative research by encouraging data integration and sharing across different institutions. AnVIL’s unique expertise has been availed for a number of high-end projects and is expected to be a focal point of many exciting new discoveries.
#9. REFLECT – A TOOL FOR OPTIMIZING COMBINATION CANCER THERAPIES

In heterogeneous diseases such as cancer, the disease progression is not the work of a single alteration but a number of co-occurring alterations requiring drugs that target all the major drivers of the disease. Through machine learning and omics data, the scientists at the University of Texas developed the platform REFLECT (REcurrent Features LEveraged for Combination Therapy) that recommends drug combinations tailored for the molecular characteristics of the tumor. The model takes in a multi-omics input of gene mutations, copy number variations, gene expression data, and protein expression aberrations of the tumor tissue to produce a personalized optimal therapy combination. The platform efficacy was validated through study data from both clinical and pre-clinical studies.
#10. FEDERATED MACHINE LEARNING POWERS RECORD-BREAKING BRAIN TUMOR STUDY

Current patient care and treatment options can gain so much from the active collaboration of patient data, but it is unfortunately not a battle easily won, and with good reasons. A hurdle that for a long time seemed impossible to surpass had been made possible by the AI approach of Federated learning. The concept of this method is to distribute the model rather than the data. The scientists at the University of Pennsylvania built the largest and one of the first federated learning models for medical data by initializing a pre-trained model with data at hand, which was then optimized by distributing the model to 71 locations across six continents. However, the model enjoyed the benefit of increased data incorporation, and no patient data was displaced from their original locations. The scientists trained the model for boundary predictions in mpMRI scans of Glioblastoma, which is a rare disease, and greatly profited through this approach. The study acts as a role model in influencing large-scale collaborations through federated learning for future clinical research.
CONCLUSION
The top 10 bioinformatics discoveries of the year 2022 have significantly advanced our understanding of biological systems and processes. From identifying new genetic markers for diseases to unraveling the complexity of gene regulation, these discoveries have the potential to revolutionize healthcare, agriculture, and many other fields. The rapid pace of bioinformatics research is a testament to the power of computational tools and techniques in driving scientific progress. As we look to the future, it is clear that bioinformatics will continue to play a crucial role in unlocking the mysteries of life and paving the way for new and innovative solutions to some of the world’s most pressing challenges.
Note: This is a list of the most exciting bioinformatics discoveries, this is not a ranking article.
Article Sources:
Nurk, S., Koren, S., Rhie, A., Rautiainen, M., Bzikadze, A. V., Mikheenko, A., … & Phillippy, A. M. (2022). The complete sequence of a human genome. Science, 376(6588), 44-53. Article
Scientists Revealed the First Complete, Gapless Human Genome Sequence ‘T2T-CHM13’. Article
Fastest DNA Sequencing that Uses AI and Accelerated Computing Sets Guinness World Record For Completing DNA Sequencing in Just Five Hours and Two Minutes. Article
Meta’s AI Protein Structure Prediction Model ‘ESMFold’ Predicts as Accurately and 6x Faster than AlphaFold2. Article
Deepmind’s AlphaFold Predicted the Structures of all the Proteins Known to Science, Expanding the AlphaFold DB by Over 200x. Article
Simulation of a Living Cell Enabled with NVIDIA GPUs. Article
Müller Paul, H., Istanto, D.D., Heldenbrand, J. et al. CROPSR: an automated platform for complex genome-wide CRISPR gRNA design and validation. BMC Bioinformatics 23, 74 (2022). Article
Wohns, A. W., Wong, Y., Jeffery, B., Akbari, A., Mallick, S., Pinhasi, R., … & McVean, G. (2022). A unified genealogy of modern and ancient genomes. Science, 375(6583), eabi8264. Article
Schatz, M. C., Philippakis, A. A., Afgan, E., Banks, E., Carey, V. J., Carroll, R. J., … & Walker, J. (2022). Inverting the model of genomics data sharing with the NHGRI Genomic Data Science Analysis, Visualization, and Informatics Lab-space. Cell genomics, 2(1), 100085. Article
Li, X., Dowling, E. K., Yan, G., Dereli, Z., Bozorgui, B., Imanirad, P., … & Korkut, A. (2022). Precision Combination Therapies Based on Recurrent Oncogenic Coalterations. Cancer Discovery, 12(6), 1542-1559. Article
Federated Machine Learning Powers the Biggest Brain Tumor Study to Date while Protecting Patient’s Data. Article
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Catherene Tomy is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She has a master’s degree in Molecular Medicine from Amrita University with research experience in the fields of bioinformatics, cell biology, and molecular biology. She loves to pull apart complex concepts and weave a story around them.
.
-
Catherene Tomy
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar
-
Dr. Tamanna Anwar





{kind=link}