
Intercellular networks between key players in the tumor microenvironments make tumor cells invincible against the body’s natural defense mechanisms. Identifying and targeting these interactions can make tumors more susceptible to tumor invasions and their eventual retardation. This study demonstrates the use of Bayesian network models in designing a network that links together these interactions by incorporating data from both single-cell and bulk RNA-seq.
For a heterogeneous ecosystem such as a tumor microenvironment, the constituents that make it up and the interactions that drive its pathogenesis and progression are highly diverse. This diversity imparts the uniqueness of each tumor in its characteristic progression patterns, immune evasion strategies, and treatment responses. This makes it crucial to identify the said interactions that capture the core processes that underlie each individual tumor to tailor treatment regimens that counter it effectively.
Having realized this need, scientists have put forward a unique workflow for uncovering the precise intercellular interactions (ICN) within the tumor microenvironments that imparts them the ability to evade immune invasions. The study was performed by intricately stringing together transcriptomic data from single-cell RNA-Seq and bulk RNA-Seq with head and neck squamous cell carcinoma (HNSCC) as the cancer of choice. The single-cell RNA-Seq was used to identify varied transcriptomic profiles within cell populations, whereas bulk RNA-Seq extrapolated that into identifying how these within-cell variations influenced the interactions between different cell populations.
DATA EXTRACTION FROM SINGLE-CELL AND BULK RNA-Seq
The data for single-cell RNA-Seq was obtained from 18 HNSCC tumors from which four cell subsets were deemed important for the current study: immune-related cells, epithelial cells, fibroblast cells, and endothelial cells. GEMs or gene expression modules are co-expressed gene cohorts, where different GEMs under the same cell subpopulations identify differences in transcriptomic processes. These transcriptomic processes could be co-evolved to accommodate the differences in intercellular communications that are sustained within individual tumor tissues. To study how the GEMs of one cell type communicate and influence the transcriptomic pattern variation of other cell types, the bulk RNA-Seq from 522 HNSCC samples was obtained from the TCGA database. The bulk RNA-Seq captures the collective transcriptomic profile of tumor samples, and it was assessed to study the influence of each GEM by GSVA scores and each cell subtype by the deconvolution method.
Gene set variation analysis (GSVA) scores can be employed as estimates of the expression of each GEM and hence the transcription processes they represent within each of the bulk RNA-Seq samples. Consensus clustering based on their GSVA scores groups together samples with a similar composition of GSVA scores, hence similar GEM expression status and hence similar transcriptomic profiles adopted by cell types. Samples clustering was also performed from the cell subtype population within them. Both clusterings resulted in four clusters with significant overlap between the two. Further support for the immune basis was provided by the differences in survival outcomes of the clusters.
MODELLING INTRACELLULAR NETWORKS
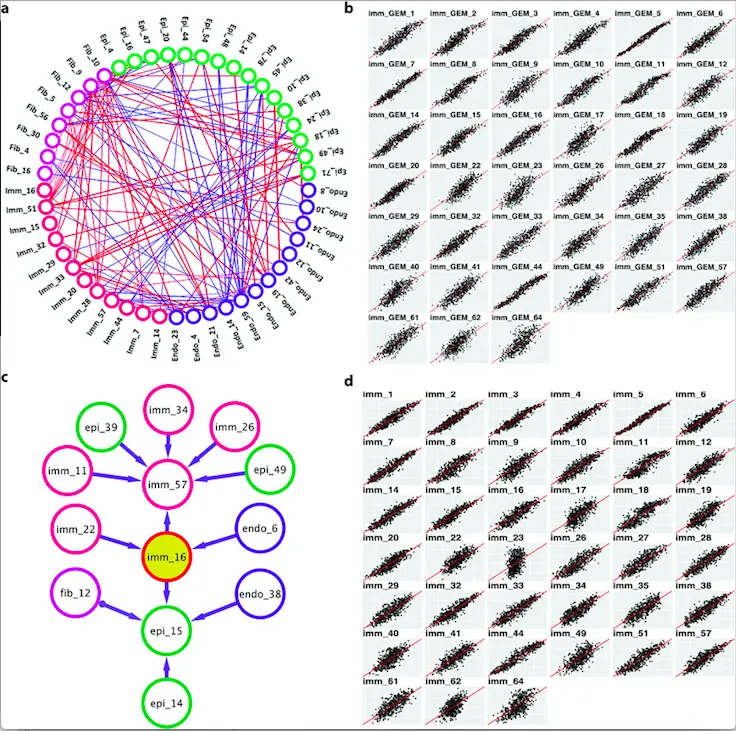
Image Source: https://doi.org/10.1371/journal.pcbi.1010761
To model the ICNs, the authors used a greedy fast causal inference algorithm (GFCI) that learns casual relationships existing within observational data. GFCI takes the GSVA scores as input and outputs a partial ancestor graph (PAG) which is a set of networks that connects together GEMs to demonstrate their influence on each other. To focus the study on intercellular networks, GFCI was used to address the intracellular PAGs, which were eliminated from the networks developed for bulk RNA-Seq. The thus remaining network is a suitable web for identifying how each cellular subtype at each transcriptomic profile (GEM) directly and indirectly influences the transcription processes of other cell types.
Further verification of the developed networks was analyzed by training regression models. From the developed network, Markov boundary GEMs for each immune GEM were identified, which were used to predict the GSVA score of that particular immune GEM. The study brought about accuracy values ranging from 0.34 to 0.85, with a mean accuracy of 0.63. This conferred that the PAG networks were able to capture and model communications between intercellular GEMs.
An in-depth analysis was applied to capture ICNs within each individual tumor and to study their deviation from the more generalized network applied above. The authors developed an individualized GFCI (iGFCI) algorithm to model this. The so developed models had 106 edges in common with the previously developed out of the 549 ones that represented the cumulation of all the ICNs, which implies it was able to capture individual characteristics of each tumor that promote immune evasiveness in them. By developing a dataset based on the presence or absence of an edge within the individual ICNs, four clusters of samples were derived. The four clusters showed different survival outcomes despite having similar clinical characteristics. Clustering patterns were also similar to ones obtained from clustering on the basis of immune GEMs, which can indicate that distinct ICNs were able to capture the distinct immune mechanisms within each cell subtype.
CONCLUSION
Immune cells are the first responders in any malfunction and are highly efficient in running our well-oiled machine, which is the human body. Though in many cases of tumor invasions, the characteristic roles of immune cells are nullified through complex networks of communications among the participating cell subtype in the tumor microenvironment. Furthermore, this diversity is customized to each individual tumor, the understanding of which can give better insights into the possible therapeutic targets for tumor retardation. This study demonstrates a successful attempt to identify and model the ICNs within HNSCC tumor samples and verify their importance through the role it plays in patient survival. This methodology can be applied to other tumor types and be a foundation to thwart tumor-sustaining strategies.
Article Source: Paper Reference
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Catherene Tomy is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She has a master’s degree in Molecular Medicine from Amrita University with research experience in the fields of bioinformatics, cell biology, and molecular biology. She loves to pull apart complex concepts and weave a story around them.
.





{kind=link}