

An international group of scientists has finally published the whole human genome sequence. The new reference genome fills in gaps left by previous versions, allowing researchers to understand better genetic variation and how it can contribute to disease in specific cases. The complete human genome sequence has already provided new perspectives to genome biology and the next decade of discoveries will reveal more about the previously unknown regions.
The Telomere-to-Telomere (T2T) Consortium sets forth a total 3.055 billion-base pair sequence of a human genome, T2T-CHM13, that involves gapless assemblies for all chromosomes except Y, improving errors in the earlier references, and presents almost 200 million base pairs of a sequence consisting of 1956 gene predictions, 99 of which are anticipated to be protein-coding.
The finished regions incorporate all centromeric satellite arrays, recent segmental duplications, and the short arms of every one of the five acrocentric chromosomes, opening these intricate regions of the genome to further variational and functional studies.
Cell Line and Sequencing
As with many earlier reference genome improvement efforts, a complete hydatidiform mole (CHM) was selected for sequencing. Most CHM genomes emerge from the deficiency of the maternal complement and duplication of the paternal complement postfertilization and are, in this way, homozygous with a 46, XX karyotype. Moreover, the sequencing of CHM13 affirmed nearly uniform homozygosity, except for a couple of thousand heterozygous variants. The local ancestry analysis revealed that most of the CHM13 genome is of European origin, including locales of Neanderthal introgression, with some anticipated admixture.
Two new DNA sequencing technologies arose throughout the past ten years that created longer sequence reads significantly. The Oxford Nanopore DNA sequencing strategy can read up to 1 million DNA letters in a solitary read with modest accuracy, while the PacBio HiFi DNA sequencing technique can learn about 20,000 letters with almost amazing precision. Researchers in the Telomere-to-Telomere consortium utilized both DNA sequencing techniques to generate the complete human genome sequence.
Genome and rDNA Assembly
The foundation for the T2T-CHM13 assembly is a high-resolution assembly string graph created directly from HiFi reads. Nodes indicate unambiguously assembled sequences in a bidirected string graph, while edges reflect overlaps between them due to repeats or actual adjacencies in the underlying genome. The CHM13 graph was created using a custom technique that blends components from previous assemblers with specialized graph processing.
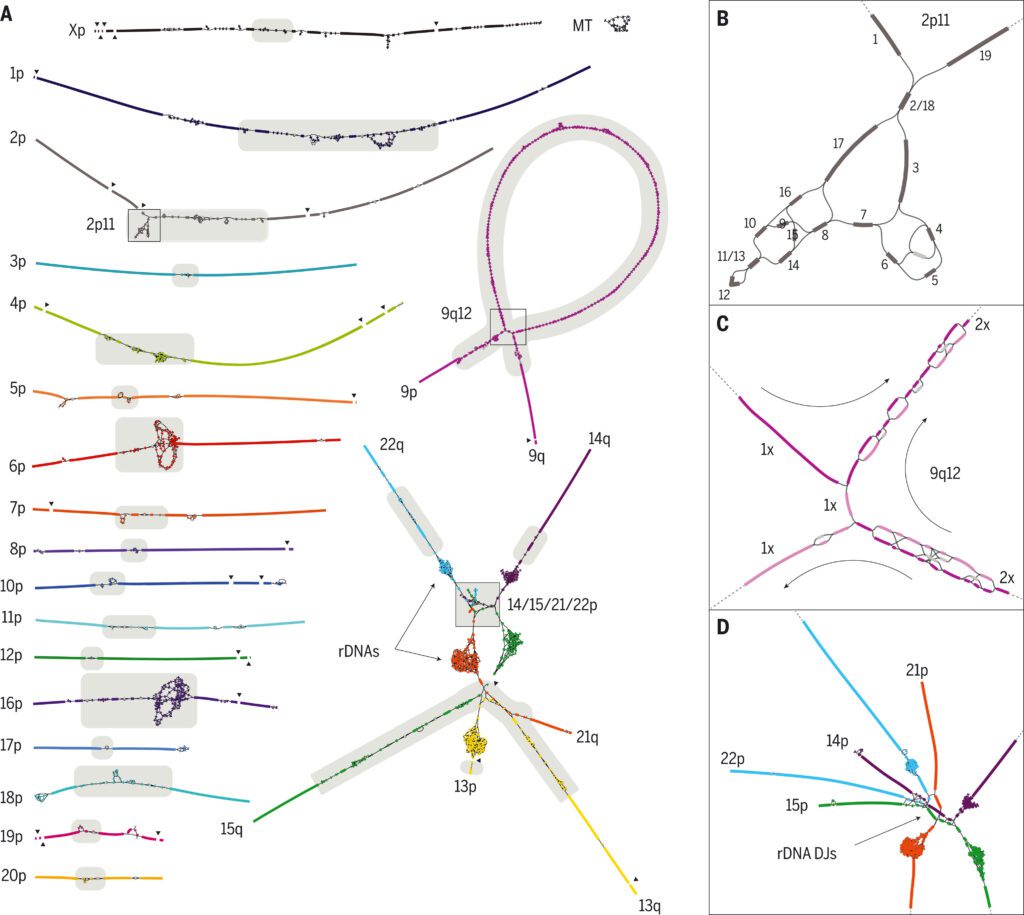
Image Source: The complete sequence of a human genome.
Most of the components in the resulting graph come from a single chromosome and have an almost linear form, implying that there are few perfect repetitions larger than around 10 kbp between separate chromosomes or distant locations.
Each chromosome’s whole sequence should be represented as a walk through a string graph, with some nodes traversed several times (repeats) and others not at all (errors and heterozygous variants). The coverage depth and multiplicity of the nodes were evaluated to help identify the right walks, which allowed most tangles to be manually resolved as unique walks visiting each node the required number of times.
The human rDNA arrays and their surrounding sequences are the most complicated part of the CHM13 string graph. Human rDNAs are 45-kbp near-identical repeats that encode the 45S rRNA and are found in huge, tandem repeat arrays within the acrocentric chromosomes’ short arms.
"A typical diploid human genome is known to have a normal of 315 rDNA duplicates, with a standard deviation of 104 duplicates."
To assemble those exceptionally unique, highly dynamic regions of the genome and conquer constraints of the string graph construction, the scientists developed Sparse de Bruijn graphs, and ONT reads were aligned to the graph to recognize a set of walks, which were changed over to sequence, fragmented into individual rDNA units, and clustered into “morphs” as per their sequence similarity.
Assembly Validation and Polishing
To assess concordance between the reads and the assembly, the scientists mapped all the available, essential primary data, including HiFi, ONT, ILMN, Strand-seq, and Hi-C-to, the CHM13v0.9 draft assembly.
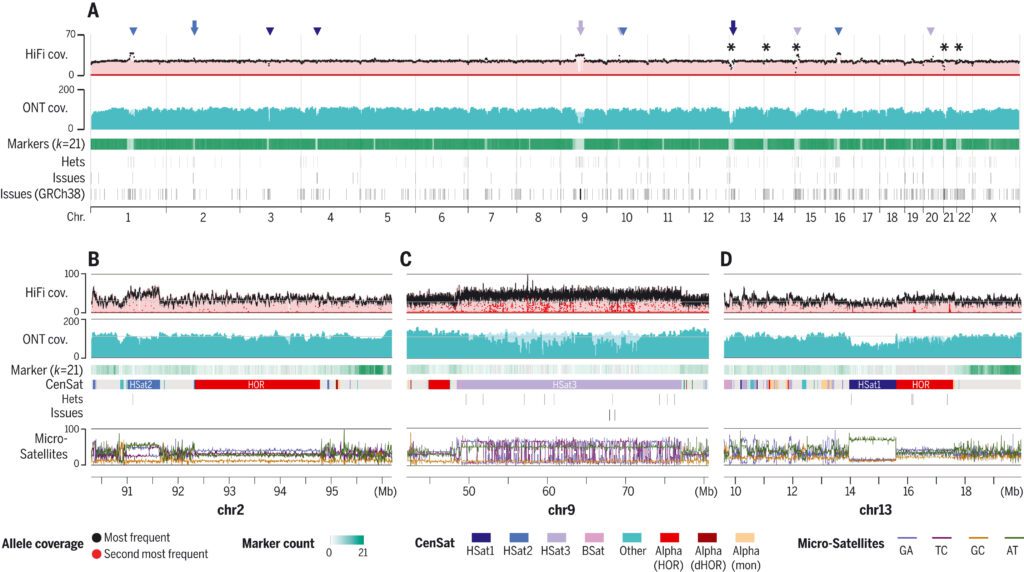
Image Source: The complete sequence of a human genome.
Manual curation amended four huge and 993 minor errors, resulting in a complete telomere-to-telomere assembly of a human genome, T2T-CHM13 v1.1.0 assembly, and recognized 44 huge and 3901 slight heterozygous variations.
Mapped sequencing read depth reveals uniform coverage across all chromosomes, with 99.86 percent of the assembly falling within three standard deviations of the mean coverage for either HiFi or ONT (excluding the mitochondrial genome) (HiFi coverage 34.70 7.03 and ONT coverage 116.16 16.96).
Since long tandem repeats are the most troublesome regions of the genome to collect, the scientists performed targeted validation of long tandem repeats to distinguish any errors missed by the genome-wide methodology.
The assembled rDNA morphs, being just of size 45 kbp each, were manually validated by examination of the read alignments utilized for polishing.
The consensus correctness of the T2T-CHM13 assembly is expected to be around one mistake per 10 Mbp, which is orders of magnitude better than the old benchmark of the “completed” sequence. Compared to GRCh38, the total number of bases covered by potential issues in the T2T-CHM13 assembly is only 0.3 percent of the total assembly length.
A Truly Complete Genome
T2T-CHM13 incorporates gapless telomere-to-telomere assemblies for each of the 22 human autosomes and chromosome X, consisting of 3,054,815,472 bp of nuclear DNA, in addition to a 16,569-bp mitochondrial genome.
This assembly adds or amends 238 Mbp of sequence that doesn’t collinearly align to GRCh38 over a 1-Mbp stretch (i.e., is non-syntenic), fundamentally containing centromeric satellites (76%), non-satellite segmental duplications (19%), and rDNAs (4%). There are no primary alignments to GRCh38 in 182 Mbp of sequence, and it is unique to T2T-CHM13. T2T-CHM13 expands the number of known genes and repetitions in the human genome.
There are 63,494 genes and 233,615 transcripts in the draft T2T-CHM13 annotation, with 19,969 genes (86,245 transcripts) projected to be protein-coding and 683 predicted frameshifts in 385 genes (469 transcripts).
Of all genes exclusive to CHM13, 140 are likely to be protein-coding on the basis of their GENCODE paralogs and have a mean of 99.5% nucleotide and 98.7% amino acid identity to their most similar GRCh38 copy. Based on their GENCODE paralogs, 140 genes distinct to CHM13 are projected to be protein-coding, with a mean of 99.5 percent nucleotide and 98.7 percent amino acid identity to their most similar GRCh38 copy.
"T2T-CHM13 highlights the genomic structure of the short arms of the five acrocentric chromosomes."
T2T-CHM13 reveals the genomic structure of the five acrocentric chromosomes’ short arms, which have mainly remained unsequenced despite their importance for cellular function. Their enrichment for satellite repeats and segmental duplications, which prevented sequence assembly and limited their characterization to cytogenetics, restriction mapping, and BAC sequencing, has resulted in this omission.
Each of the five of CHM13’s short arms follows a similar design which comprises an rDNA array embedded within distal and proximal repeat arrays.
The sizes of those repeat arrays differ among chromosomes, yet despite their variability in size, all satellite arrays share a serious level of similitude (typically >90% identity) both between acrocentric chromosomes and between acrocentrics, setting out many open doors for interchromosomal exchange and non-heterogeneous recombination.
Future Prospects of the Now-Complete Human Genome Sequence
The T2T-CHM13 assembly contributes five full chromosome arms and more additional sequence than any genome reference release in the past 20 years. The 8% of the genome has been disregarded not because it is unimportant but because of technological constraints. Long-read sequencing with high precision has now broken down this technological barrier, allowing for comprehensive analyses of genomic variation across the whole human genome, which will drive future discoveries in human genomic health and disease.
The now-complete human genome sequence will be especially significant for studies that expect to lay out far-reaching perspectives on human genomic variation or how individuals’ DNA contrasts. Such knowledge is fundamental for getting the genetic contributions to specific illnesses and for utilizing genome sequence as a routine piece of clinical care later on. Many research teams have proactively begun using a pre-release adaptation of the complete human genome sequence for their research.
Producing a really complete human genome sequence addresses an incredible scientific accomplishment, giving the main comprehensive perspective on our DNA blueprint. This foundational data would reinforce the numerous continuous endeavors to see each of the functional distinctions of the human genome, which thus would enable genetic studies of human infection.
The complete sequencing expands upon being crafted by the Human Genome Project, which mapped around 92% of the genome, and research embraced from that point forward. A vast number of specialists have grown better research center apparatuses, computational techniques, and key ways to deal with unraveling the intricate sequence.
That last 8% incorporates various genes and redundant DNA and is equivalent in size to a whole chromosome. Scientists created the whole genome sequence utilizing a human cell line with just a single duplicate of every chromosome, not at all like most human cells, which convey two duplicates of every chromosome. The researchers noticed that the more significant part of the recently added DNA sequences was close to the repetitive telomeres (long, trailing ends of every chromosome) and centromeres (thick center areas of every chromosome).
"In the future, when someone has their genome sequenced, we will be able to identify all of the variants in their DNA and use that information to better guide their healthcare. Truly finishing the human genome sequence was like putting on a new pair of glasses. Now that we can clearly see everything, we are one step closer to understanding what it all means.” T2T consortium co-chair Adam Phillippy, Ph.D.
The complete genome sequence analysis would add heaps to our knowledge base of chromosomes, including higher accurate maps for five chromosome arms, which opens new areas and opportunities for research. This helps answer fundamental science questions regarding how chromosomes appropriately segregate and divide.
Story Source: Nurk, S., Koren, S., Rhie, A., Rautiainen, M., Bzikadze, A. V., Mikheenko, A., … & Phillippy, A. M. (2022). The complete sequence of a human genome. Science, 376(6588), 44-53. https://www.genome.gov/news/news-release/researchers-generate-the-first-complete-gapless-sequence-of-a-human-genome
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}