
Researchers at Meta AI developed an artificial intelligence (AI) model that can precisely predict entire atomic protein structures from a single sequence of a protein. The use of language model representations to replace the requirement for explicit homologous sequences distinguishes ESMFold from AlphaFold2. As a result of this simplification, ESMFold is significantly faster than the MSA-based tools.
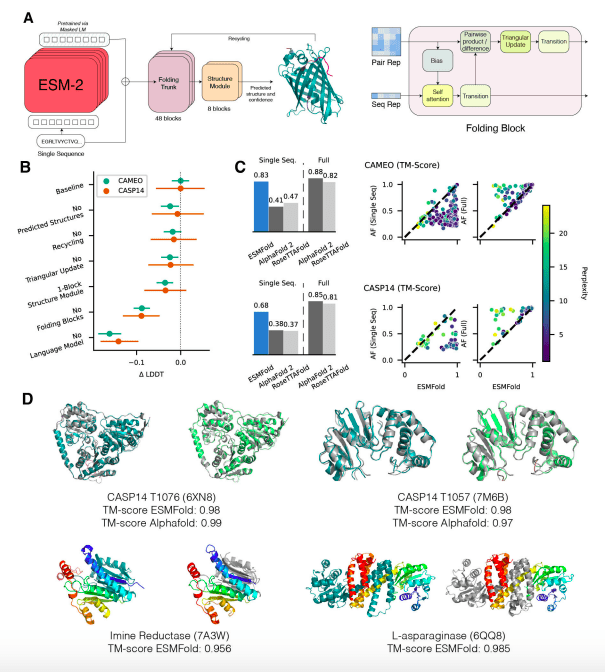
Image Source: https://doi.org/10.1101/2022.07.20.500902
A recent study found that large language models develop emergent capabilities over time, going beyond pattern matching to generate lifelike images and text and perform higher-level reasoning. There is little information about what language models can learn about biology as they are scaled up from smaller studies involving protein sequences.
What is ESMFold?
ESMFold is a protein structure prediction model which uses transformer models to encode protein sequences. It is significantly faster than other models which use multiple sequence analysis (MSA) yet still maintains high-quality predictions. Researchers can also integrate the ESM (Evolutionary Scale Modeling) project’s own MSA model in conjunction with ESMFold in order to increase the accuracy of predictions.
To date, this study has been the largest language model evaluation of proteins with 15 billion parameters. Scaling models uncover information that enables the prediction of three-dimensional protein structures at individual atom resolution as they learn information. An atomic level structure prediction system based on protein sequences is presented, which provides high accuracy end-to-end atomic level predictions. As long as the language model understands the sequences, ESMFold’s accuracy is similar to AlphaFold2 and RoseTTAFold. A practical timescale for the exploration of metagenomic protein structures can be achieved with ESMFold inference, which is an order of magnitude faster than AlphaFold2.
By constraining the pattern of words in a text, the distributional hypothesis suggests that meaning can be inferred. Sequence inference in biology has been based on an analogous idea. Biological structure and function can also be inferred from sequence patterns since mutations selected through evolution are constrained by the structure and function of a protein. This would provide insight into some of biology’s most fundamental problems.
In spite of this, getting sufficient information about biological structures and functions from sequence alone remains difficult. As computing data, and model size increase, general-purpose language models perform better on complex tasks in natural language processing and artificial intelligence. Scaling up a simple training process to large corpora of data leads to useful capabilities for language models. Examples include few-shot translation, common-sense reasoning, and mathematical reasoning.
By taking advantage of the inference time advantage, large metagenomic sequence databases can be efficiently mapped structurally. A rapid and accurate structure prediction tool like ESMFold is excellent for identifying remote homology and conservation in a large collection of novel sequences. The ability to predict millions of protein structures within practical timescales can lead to new insights into the diversity and breadth of natural proteins as well as the discovery of new protein structures and functions.
In order to accurately predict protein structure at the atomic level, large protein language models need to learn a sufficient amount of information. The ESM-2 language model is the largest language model of protein sequences to date, with variations that can reach 15 billion parameters. With ESMFold, one can predict end-to-end 3D protein structures based on only a single sequence using the information and representations learned by ESM-2. This also allows us to quantify the emergence of protein structure as the language model grows from millions to billions of parameters. The accuracy of structure prediction increases as the size of the language model increases.
In order to achieve optimal performance, AlphaFold2 and RoseTTAFold use multiple sequence alignments (MSAs) and templates of similar protein structures in order to achieve breakthrough success in atomic-resolution structure prediction.
ESMFold, on the other hand, generates structure predictions using only one sequence as input by leveraging the internal representations of the language model. When given a single sequence as input, ESMFold produces more accurate atomic-level predictions than AlphaFold2 or RoseTTAFold and competes with RoseTTAFold when given full MSAs.
As a result, ESMFold produces comparable predictions for low-perplexity sequences as well as that structure prediction accuracy correlates with language model perplexity in general, suggesting that when a language model is better able to comprehend a sequence, it is also better able to comprehend a structure.
A major advantage of ESMFold is its exponentially faster prediction speed than existing atomic-resolution structure predictors. This will allow it to bridge the gap between the rapid growth of protein sequence databases containing billions of sequences and the slower development of protein structure and function databases. ESMFold is used to rapidly compute one million predicted structures, representing a diverse subset of metagenomic sequence spaces that lack annotated structure or function.
ESMFold’s high-confidence predictions have little similarity to any known experimental structures, which suggests metagenomic proteins may be structurally novel. There is a high degree of sequence similarity between many high-confidence structures and UniRef90 entries, indicating generalization of the model’s predictions beyond the training dataset and providing structure-based insight into protein function not available from sequence information alone. As a result of exploiting ESM-2’s unprecedented view of protein sequence language, ESMFold will enhance our understanding of large databases of poorly understood protein sequences.
With just an order of magnitude fewer parameters than the largest models of text developed recently, the ESM-2 family of models is the largest protein-language model developed to date. In addition to its significant improvements over predecessor models, ESM-2 can capture a more accurate picture of structure even at 150M parameters than the language models of the ESM-1 generation at 650M parameters.
Protein-language models like ESM-2 are regarded favorably by other recent, significant models. The language model is ESMFold’s biggest performance driver. When ESM-2 understands the sequence well, we can obtain predictions that are comparable to those made by other models when language modeling perplexity is high. It is possible to obtain accurate atomic-resolution structure predictions with ESMFold, which is up to two orders of magnitude faster than AlphaFold2.
Final Thoughts
Language models with unsupervised learning objectives enable atomic-resolution prediction of protein structures. Scaling language models up to 15 billion parameters enables a systematic study of the effect of scale on the learning of protein structures. There is a strong link between how well the language model understands a sequence and the structure prediction that emerges. ESMFold has similar accuracy to AlphaFold2 and RoseTTAFold for sequences with low perplexity that are well understood by the language model.
Article Source: Reference Paper | ESMFold Code: GitHub Repository
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Srishti Sharma is a consulting Scientific Content Writing Intern at CBIRT. She's currently pursuing M. Tech in Biotechnology from Jaypee Institute of Information Technology. Aspiring researcher, passionate and curious about exploring new scientific methods and scientific writing.











{kind=link}