
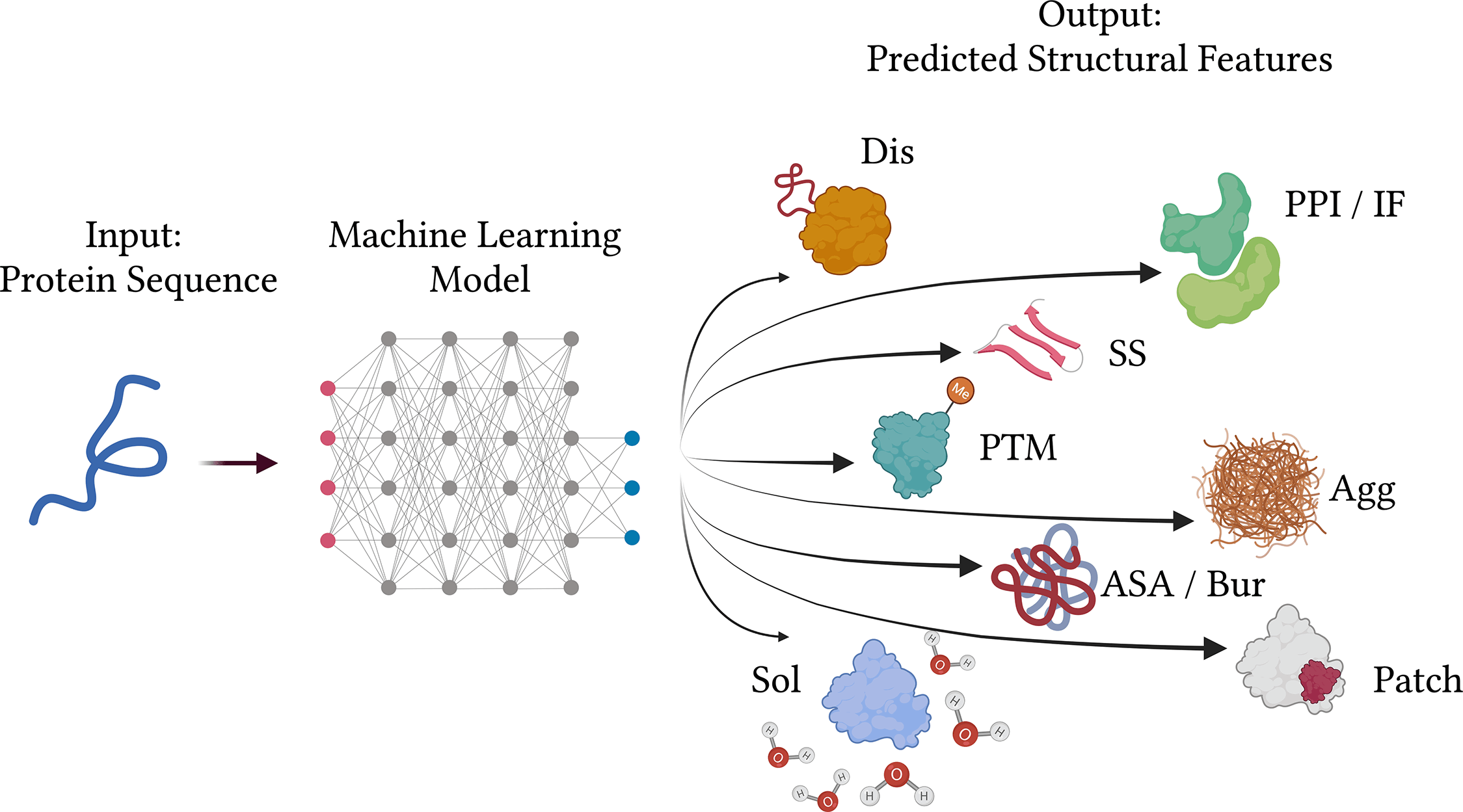
The success of machine learning-based approaches for the prediction of protein properties from their amino acid sequences can be attributed to the overall accessibility of genome sequencing data. Several reoccurring difficulties have been noted throughout time while reading submitted manuscripts and released publications, which makes it difficult to comprehend and reproduce certain stated findings.
A legitimate worry may be that machine learning specialists don’t have all the information necessary to apply their techniques to proteins properly or that biologists don’t understand how it works. In this article, we try to close this gap for those who are interested in creating such approaches.
One of the most crucial challenges for bioinformaticians is still predicting the functional properties of proteins. Ten issues or guidelines that reflect best practices, especially for techniques that predict the functional, structural features of proteins using protein sequence data as input, are shown below to help developers overcome this gap:
Sequence-based approaches often perform worse than those based on structures. There has been significant development in the field of structure prediction during the past several years, which has coincided with the expansion of empirically determined structures. Particularly, AlphaFold2 represents a significant advancement in structure prediction. Many significant protein types and areas still lack access to accurate structural data, nevertheless. Furthermore, it hasn’t been shown yet and could still be fairly restricted how valuable predicted structures are as input for the prediction of functional features like interface areas or binding sites. Structure-based functional property prediction is not always more precise than sequence-based methods. In order to close this gap, the area of sequence-based prediction of protein function and structure features was created.
Countless machine learning model developers in this subject have backgrounds in either biology or computer science, and they may adopt implicit assumptions and biases from those fields without realizing that they may not be appropriate for the “other” field. Bioinformatics-trained model creators, who could be assumed to have a broader perspective, might also overlook some machine learning or protein science best practices. This article’s main objective is to provide advice that will be helpful to scientists with various backgrounds striving to create cutting-edge prediction models for proteins.
#1 Answer a biological problem you aim to solve
Choose a biological question and explain why and how the machine-learning architecture matches sequence data when proposing your machine-learning technique to predict protein properties from the sequence. If anything like syntax or grammar-like structures are relevant and to count on more, this is simply a flimsy understanding of how things may operate in practice. Protein sequences are neither pictures nor sentences in a language. The overall structure and function of the protein are supported by its specific amino acid sequence, which itself has a long evolutionary history. This would be lost in these parallels since it is quite dissimilar from pictures or phrases in a natural language. Therefore, the difficulty is to lead the reader to your machine-learning architecture from a typical prediction output for protein sequences rather than from cats or the English language. As a result, while it might be enticing to use language like a well-known comparison to assist the reader in understanding a concept, using examples from similar and well-known biological problems can help the reader better appreciate the relevance and context of the work.
#2 Give a good description of the Database
The description of the dataset should include the following information:
- The source databases utilized (such as the PDB),
- The selection criteria (such as the minimal resolution and certain experimental methodologies), and
- Applying further filtering techniques (such as those based on sequence identity,
The features are a crucial component of the dataset. These might include data directly derived from the sequences, such as the kind of amino acid, evolutionary data (like profile representations in the form of PSSMs from PSI-BLAST), or characteristics predicted by other techniques, including the secondary structure and solvent accessibility. Utilizing representation models, such as those found in transformers, features may also be learned.
#3 Minimize data leakage by excluding homologs.
Sequence redundancy must be eliminated, and homolog predictions between the test set and training set avoided by applying a filter based on sequence identity, such as any pair of sequences are not allowed to share more than 25% of the same amino acids.
#4 Define the type of input and output of the model
Sequence-based approaches need to be divided based on the sorts of input and output that are needed. Although the protein sequence is always the input, the prediction model may receive a sequence window, a single residue, or the entire protein sequence. The linked characteristics are always the model’s real input. Regular neural networks and random forests, which are often simpler approaches, typically only accept one residue or one window position as input, but deep learning models gain their power from their capacity to recognize patterns over the whole sequence.
#5 Crucial benchmarking measures
Good benchmarking is essential for introducing your prediction approach properly and, in fact, for critically evaluating it. Choosing the best benchmarking measures is crucial since different measurements emphasize different performance characteristics of your approach. A high-class imbalance is seen in many protein-specific classification tasks; for instance, only 10% to 15% of a protein’s residues interact on average. Accuracy, though frequently utilized, can be deceptive when working with severely uneven data. Since there are no true biological patterns in the data, the machine learning model in this scenario merely learns to predict the non-interacting class, which makes up the bulk of the data.
#6 – Listing out the positives and negatives in PPI interface data.
A residue that was seen to be interacting is considered positive; nonetheless, it is typically challenging to acquire negative evidence for PPI. Negative data, such as peptides that were tested and found not to bind, may be available for some portions of the protein for epitope-antibody interaction. You may choose to completely ignore buried remnants or consider them unfavorable.
#7 Bring out the correspondence in the results with respective models and training/test data
A study frequently introduces a number of models, their modifications, and accompanying datasets. For instance, you may incorporate many feature sets or architectural building pieces, each of which results in a distinct model. Determining which model was trained on which training set, which findings were dependent on which model, and which test set each model was based on is crucial when presenting the results.
#8 Building an efficient interpretation of the model
Based on data from Hou and colleagues, the example illustrates how the pattern of conservation changes across the surface (Sur), interface (Inter), and buried (Bur) residues for protein-protein interactions (PPIs) and epitope interactions, which are particular forms of PPI (bottom).
Retraining and testing after removing a particular characteristic from the input is a valuable technique to confirm its effect. It has been demonstrated that the AUC-ROC values fall noticeably, significantly, and repeatedly using the global characteristic of protein length.
#9 Providing significant estimations
Since comparison is the primary focus of benchmarking, it seems appropriate to provide a significant estimate for any difference that you want to infer is significant. You could also choose to lower the number of digits you publish, for example, in tables, based on the significance estimations.
#10 Open accessibility to your methods
Numerous publications, such as Bioinformatics, PLOS Computational Biology, and BMC Bioinformatics, currently demand that the technique be provided alongside the manuscript in some form, such as code or a web server. Your approach must be accessible in order for other researchers to utilize it, evaluate it, and carry on with their study. Naturally, your article offers a thorough explanation of how it was accomplished, but open-source code and a functional web server are also crucial. Reproducing a method from the article is time-consuming and frequently impractical. Even executing someone else’s code can be quite challenging, and many prospective users of your approach might not be tech-savvy enough to overcome these challenges.
Final thoughts on the tips
In this article, we went over 10 key issues that a developer of a sequence-based functional, structural properties prediction tool must take into consideration as they work through the various steps, from posing the biology question to preparing the datasets for training and testing to releasing the predictor to the public. There are a few guidelines that may be applied—some of them fairly obviously—to algorithms that employ protein structural characteristics as input, as well as the more broad field of machine learning.
Article Source: – Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Riya Vishwakarma is a consulting content writing intern at CBIRT. Currently, she's pursuing a Master's in Biotechnology from Govt. VYT PG Autonomous College, Chhattisgarh. With a steep inclination towards research, she is techno-savvy with a sound interest in content writing and digital handling. She has dedicated three years as a writer and gained experience in literary writing as well as counting many such years ahead.





{kind=link}