
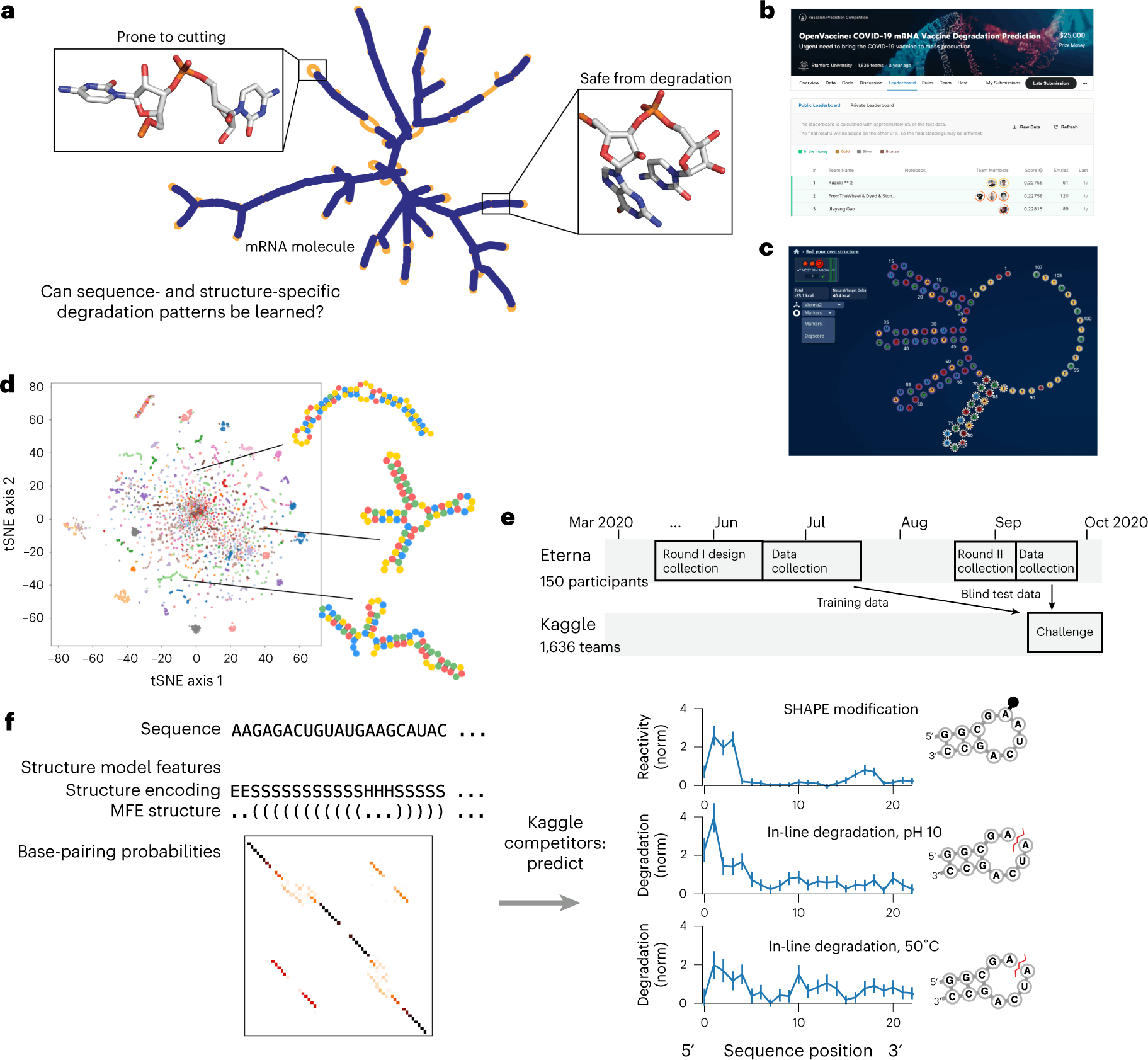
Scientists at the University of Stanford display the potential of crowdsourcing in accelerating and enhancing the research process. The study utilized two crowdsourcing platforms, Eterna and Kaggle, to design the RNA input and develop models that predict their degradation profile to aid in the design of stable mRNA therapeutics.
The intrinsic instability that causes self-destruction through an in-line hydrolysis reaction by the unstable 2’-OH attacking the phosphodiester bonds makes the widespread use of mRNA therapeutics a flight of fantasy. They reign as the superior option to their recombinant protein counterparts in many aspects: easier and faster to synthesize, far less expensive, and can be targeted at or translated as any protein of choice. But the possibilities they present are banged shut in the face of the resources and expenses needed for their transportation and storage. To salvage the integrity and concentration of the mRNA molecules, they are maintained at sub-zero temperatures that rival the Antarctic. Hence, to bring this figment of imagination to life, there exists no better solution than to develop stable mRNA molecules, a concept that is much easier in theory than in practice.
In an effort to speed up the entire process and generate fresh ideas for the field, the scientists at the University of Stanford leveraged two crowdsourcing platforms to tackle the above problem. First, Eterna’s gaming platform was used to design diverse sets of RNA molecules that were fed into a Kaggle competition: ‘Open Vaccine: COVID-19 mRNA Vaccine Degradation Prediction’. Within six months, the study managed to source robust predictive models that analyze RNA degradation to aid in the design of more durable mRNA therapeutics.
Sourcing of RNA Designs
The study was performed in two sessions. They first acquired the input data required for the predictive model through Eterna, a scientific gaming platform that allows players to design RNA molecules based on a given puzzle, which is a shape and/or a set of design patterns for the target RNA. The players are provided the freedom to construct RNA molecules within the given requirements of the puzzle. The generated designs can then be validated and improved by the feedback from scientists who create and test the designed RNAs within the labs.
Through various challenges conducted in Eterna, scientists are able to gain diverse solutions to RNA-based problems. Challenges can be posted for specific goals as the puzzles to be solved in designing the RNA molecules. This allows various labs to source hundreds and thousands of RNA designs for their studies. The Open Vaccine Challenge seeks entries for more stable mRNAs that code for the same amino acid sequence as that of the COVID-19 spike protein. The ‘Roll Your Own Structure’ was initiated as a subsidiary challenge within The Open Vaccine and compiled 3029 submissions of 68 nt long and 3005 submissions of 102 nt long in Round I and Round II, respectively.
The RNA degradation rates were determined by In-line seq, a method that determines the degradation of each nucleotide within the RNA, while the SHAPE assay was employed to study the secondary structure motifs. The degradation data was collected from RNA molecules subjected to accelerated degradation conditions: 10mM magnesium with a pH of 10 and 10mM magnesium with a temperature of 50°C. The data from the above two states, along with the SHAPE assay, acted as the target variables to be predicted for each nucleotide within the RNA. Input features for the model building included data on RNA sequences and their secondary structures. The accuracy of the model is determined by the error measure, mean column RMSE (MCRMSE).
Sourcing of Degradation Model
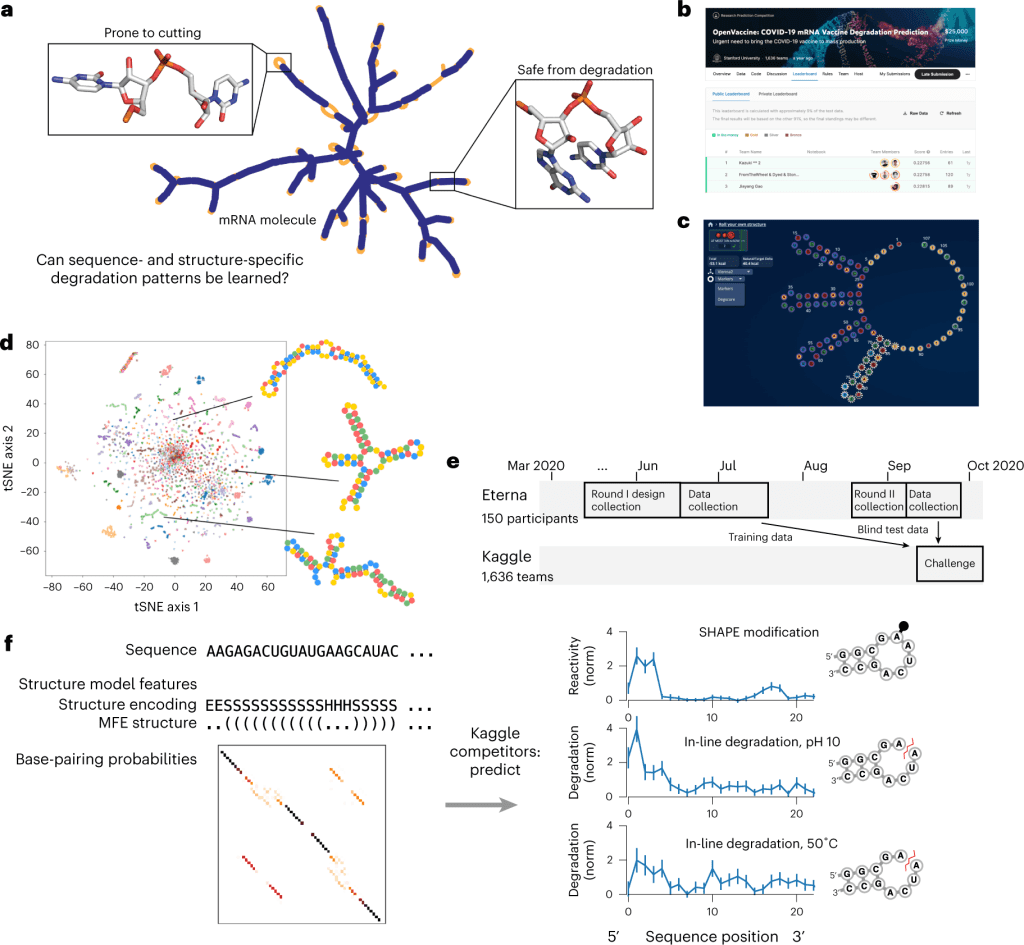
Image source: https://doi.org/10.1038/s42256-022-00571-8
The models were trained on the 3029 RNA structures from ‘Roll Your Own Structure’ round I, where 629 structures were split into the public test set while the rest 2400 were used as the training set. The final scoring is performed through blind evaluation on the private dataset that contains the 3005 structures from round II, which helps assess robustness against overfitting. The longer RNA molecules of the private dataset, as compared to the public dataset, help study the model generalizability for longer RNA sequences.
The best models were based on their performance on the private dataset. The top models from the Kaggle competition, Nullrecurrent, and Kazuki2 produced MCRMSE of 0.22758 and 0.22756 on the public dataset and 0.34198 and 0.34266 on the private dataset, respectively. A comparison between the Kaggle models and the reference predictive models, DegScore and DegScore-XGB, showed an improved predictive accuracy of 37% on the public dataset and 25% on the private dataset. In addition to the private dataset, the top two models were also tasked with predicting the degradation of full-length mRNA sequences that have a median length of 928, about ten folds longer than the ones in the training set. Both models displayed better performance than the references, with the Kazuki2 model having the highest correlation of 0.48 with the experimentally assessed degradation values.
Future Directions
The model’s ability to capture structural motifs and predict degradation values for individual nucleotides with low error profiles could deliver invaluable assistance in designing mRNA molecules with the desired attributes for high stability. Aggregation of the two top models by ensemble methods only displayed modest improvement in the predictive power, indicating that the models were able to capture the majority of signals from the input features. The authors suggest further advancements for the current study through natural language processing models that could tackle text generation approaches and models that can handle modified bases such as pseudouridine.
Article Source: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Catherene Tomy is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She has a master’s degree in Molecular Medicine from Amrita University with research experience in the fields of bioinformatics, cell biology, and molecular biology. She loves to pull apart complex concepts and weave a story around them.
.





{kind=link}