
Single-cell RNA sequencing (scRNA-seq) has proved to be a powerful tool for deciphering the entire repertoire of transcripts in a single cell. Traditional sequencing techniques detect averaged transcript levels for cell populations. The emerging technology of scRNA-seq enables the classification of cell subpopulation and cell heterogeneity. The authors of the review focus on the recent technological advancements in the scRNA-seq field and discuss several applications for the technique, encompassing several facets of fundamental as well as medical research. They also present an outlook on the application of the technique in traditional Chinese medicine.
The significance of single-cell RNA sequencing
The determination of the transcriptome, the whole repertoire of mRNAs at a certain developmental stage of a cell, is the ultimate purpose of the scRNA-seq technique. Transcriptome sequencing involves sequencing the cDNA library of all RNAs in a cell and monitoring the overall transcriptional activity of the cell at the single nucleotide level. The technique enables the identification of novel rare and unknown transcripts as well as the analysis of structure and expression levels of the transcriptome, thereby enhancing our understanding of gene expression and regulation.
Traditional RNA sequencing methods are capable of detecting averaged gene expression levels for the cell population rather than the specific expression of cell subsets. With the advent of high-throughput sequencing technologies, the ability of RNA sequencing technologies has enhanced the detection limits of transcriptome sequencing as well as unbiased transcriptome analysis at the single-cell level.
Gene mutations and alterations, thereby resulting in changes in gene expression, can now be better understood at the single-cell level by using scRNA-seq techniques. Rare somatic mutations, including single-nucleotide variations (SNVs) and copy number variations (CNVs), are easily detectable using scRNA sequencing. Single-cell sequencing has revealed genome-wide somatic SNVs in hippocampal and prefrontal cortical neurons. Somatic SNVs have been shown to accumulate more rapidly at the onset of neurodegenerative diseases. Hence, the identification of such mutations is crucial in terms of drug design and therapeutics. Also,genome-wide single-cell sequencing has revealed somatic SNVs-rich genes in endocrine cells, which are associated with oxidative damage. Thus, scRNA-seq has remarkably enabled the identification of disease-causing mutations as well as generated transcriptome data at the single-cell level for fundamental research.
The evolution of scRNA-Seq technologies
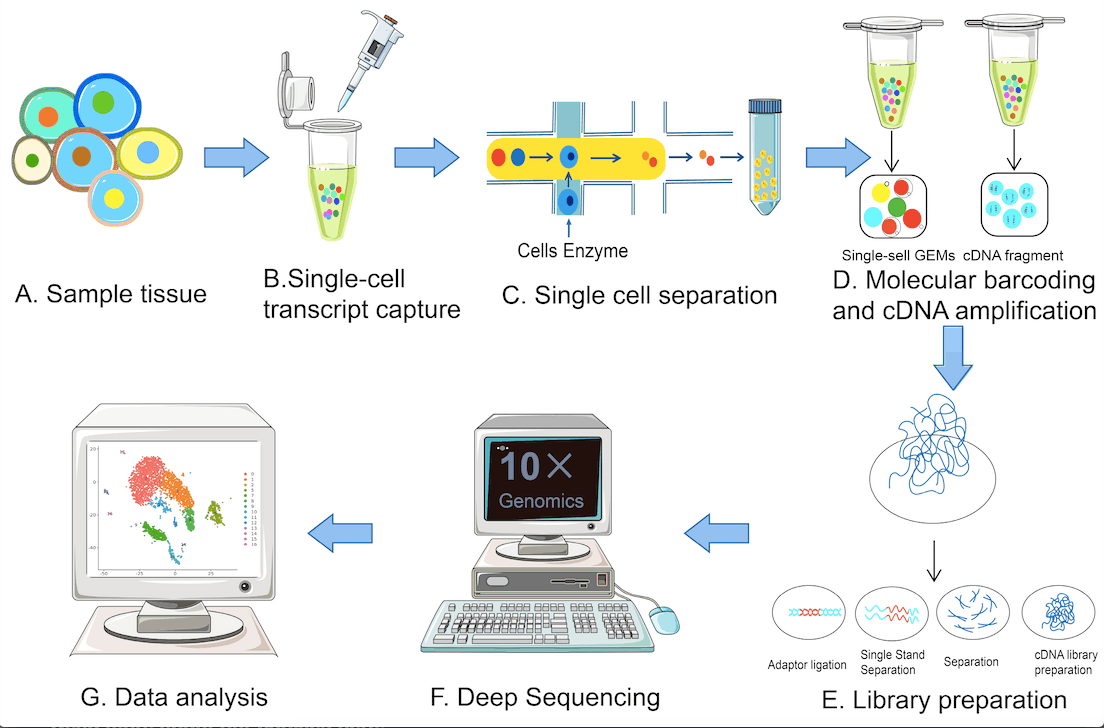
The single-cell sequencing techniques typically involve the following steps: single-cell isolation, reverse transcription, cDNA synthesis, single-cell library construction, high-throughput sequencing, and data analysis. The following figure illustrates the various steps.
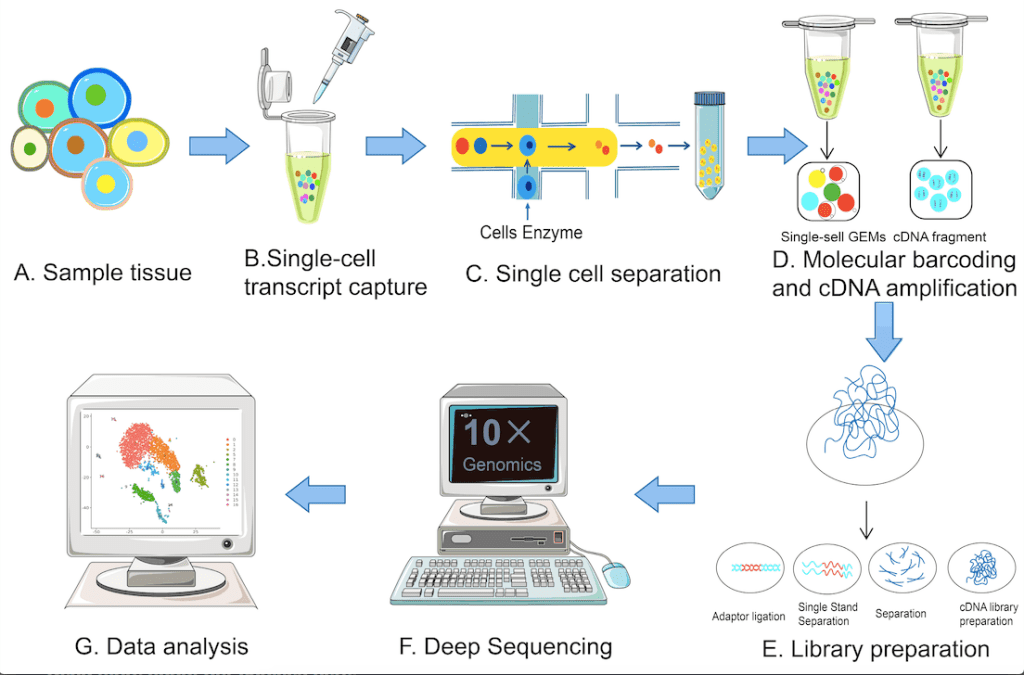
Image source: https://www.mdpi.com/1422-0067/24/3/2943#B1-ijms-24-02943
The two core aspects of scRNA-seq are obtaining single cells and the amplification of single cell-cDNA for library construction. Several innovations catering to these aspects have led to the development of a series of sequencing technologies like Smart-seq2, VASA-seq, Seq-Well, 10× Genomics, and many more.
Single-cell sorting in scRNA sequencing is achieved using microfluidics, an integrated system comprising integrated fluidic circuits (IFCs), microporous methods, and droplet methods as opposed to traditional cell sorting techniques.
The Phi29 polymerase replication method is used for the amplification step. This method, as opposed to earlier traditional methods of amplification, is seen to increase amplification efficiency and is suited for long sequence runs. The method is also capable of forming complete cDNA strands during amplification.
Among the various sequencing methods, 10× Genomics is the choice of sequencing method for the various single-cell sequencing methods being reported due to its high throughput and low cost. The VASA-seq method enables the detection of non-coding RNAs and selective promoters.
Applications of scRNA-seq in various fields
The emerging technology of single-cell sequencing has found applications in various fields, both in terms of fundamental research as well as therapeutics. Currently, it is being used in several fields such as embryonic, tissue, and organ development, tumor and immune system research, research on metabolic heterogeneity as well as in immunology.
Currently, scRNA-seq techniques are used for classifying cells at various stages of embryonic development and identifying specific markers associated with embryonic competence. The scRNA-seq atlas is a high-dimensional search space that encompasses several endodermal organs, including human respiratory as well as gastrointestinal developmental transcriptomic information.
In tumor research, scRNA-seq reveals the tumorigenesis mechanism and cell mutation process from the molecular level, which was previously unattainable. scRNA-seq has been used to classify primary breast tumors at the single-cell level, which clearly aids in the development of further cancer therapeutics.
In immunology research, scRNA-seq has been used to elucidate the heterogeneity, dynamics, and transcriptional profile of human CD4+ T cells, which reduce excessive inflammation.
The authors have also reported the significance of scRNA-seq techniques in traditional Chinese medicine (TCM) in terms of syndrome differentiation study, the efficacy of TCM, and the elucidation of toxic mechanisms of TCM.
Conclusion
The single-cell RNA sequencing technology is a revolutionary technique in the field of transcriptomics. Its applications range from understanding and identifying mechanisms at the embryonic stage up to the organ level with the aid of single-cell level information. Despite the widespread applications of the techniques, they have their own share of limitations. Firstly, the single-cell sorting results in the loss of cellular integrity as well as activity. Secondly, the high-dimensional data generation at the sequencing step results in large amounts of noise, which affects the technique’s efficacy. Cellular processes such as cell differentiation as well as division are dynamic processes, and the currently available scRNA-seq methods do not incorporate this. Nevertheless, the technology has proven to be of huge benefit in terms of determining transcriptomic data at the single-cell level, thereby aiding basic research as well as therapeutics.
Article Source: Reference Paper
Learn More:




Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}