
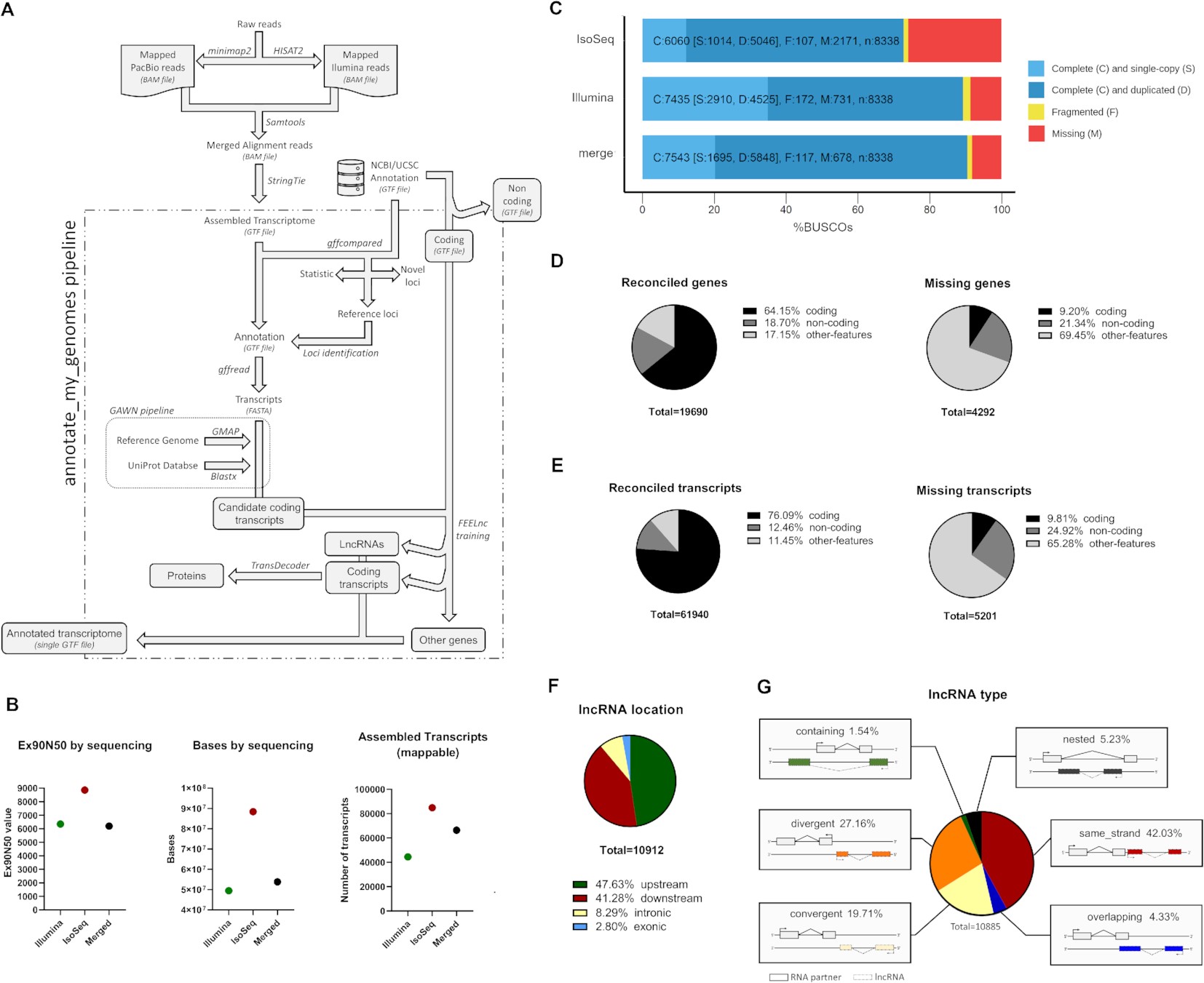
A simple genome-guided transcriptome annotation pipeline created by researchers from Chile and the University of Manitoba in Winnipeg, Manitoba, Canada, that takes compiled transcripts from hybrid sequencing information as input and recognizes coding and long non-coding RNAs by integrating a number of bioinformatic techniques, including gene reconciliation with earlier annotations in GTF format.
The expansion of genome assemblies, which are frequently annotated using hybrid sequencing transcriptomics, is a result of the development of hybrid sequencing technologies. This has a positive impact on genome characterization and the discovery of novel genes and isoforms in a range of organisms.
The current study introduces annotate-my-genome, a simplified transcriptome annotation pipeline that takes as input assembled transcripts from hybrid sequencing data and distinguishes between coding and long non-coding RNAs (lncRNAs) by integrating a number of tried-and-true methods, such as gene integration with earlier annotations. Comparing this technique to more conventional Illumina or PacBio RNA-seq methods like IsoSeq, as well as related pipelines, it produces superior transcriptome assembly and annotation.
Annotate-my-genome: Enhancing Transcriptome assemblies
New genome assembly techniques and the emerging technology of Next Generation Sequencing (NGS) have significantly enhanced genome characterization, revealing novel genes and isoforms in both model and non-model species. The resolution of transcriptomes using RNA-sequencing (RNA-seq) based on short reads is constrained by the assembly process’s technological constraints. The quality and coherence of transcriptome assemblies are frequently enhanced by long-read RNA-seq methods, either by itself or in combination with short-read sequencing. When using error correction and polishing pipelines, long-read technologies like PacBio single-molecule real-time (SMRT) and Oxford Nanopore (ONT) sequencing technologies (hereafter PacBio and Nanopore sequencing, in both) are more effective than short-read RNA-seq at reconstructing whole transcripts. IsoSeq and IsoCon, two well-known PacBio-only based pipelines, frequently excel at these tasks, and hybrid sequencing even exceeds these approaches, creating superior transcriptome assemblies.
Although comprehensive and trustworthy genome annotations are provided by evidence-based proteomics and transcriptomics for genetic analysis, specialized tools for hybrid RNA-seq analysis are also required.
Data gathering: Illumina(short-reads) and PacBio (long-reads)
To get the samples, RNA-sequencing (Transcriptome) datasets of the chick embryo’s subcommissural organ (SCO) were collected. In outbred Gallus gallus embryos, 25 SCOs were dissected at Hamburger-Hamilton (HH) stages HH23 and HH30. The efficiency of this method is demonstrated by accurately assembling the SSPO gene in chickens, which has more than 105 exons, and by using homology assignments to map missing genes in the chicken genome sequence. Notably, the data reported here presents the first transcriptional profiles of the subcommissural organ (SCO) of the chick embryo, a brain gland associated with many morphogenetic processes, including the control of brain development and body axis alignment.
Assessing RNA-sequencing Assembly
Augmentation of the chicken transcriptome’s gene annotation by combined PacBio and Illumina RNA sequencing assembly.
The objective is to use the FEELnc classification tool to categorize the reported long non-coding RNAs (lncRNAs) by location and subtype. The final results proved that hybrid sequencing is advantageous for a thorough and consistent characterization of a particular transcription. Additionally, this technology offers a simple approach to streamline the annotating process conveniently.
The hybrid sequencing technique allows for a complete reconstruction of the Giant SSPO gene.
The primarily secreted glycoprotein that makes up the Reissner fiber from the subcommissural organ is encoded by the massive gene SSPO, which has 105 exons. The pipeline’s purpose was to construct and annotate this gene. In order to test our hybrid alignment assembly and characterization workflow, we used the chicken SSPO gene, which the NCBI has provisionally designated as a protein-coding gene. In conclusion, hybrid sequencing improves contiguity and decreases disassemblies by incorporating advanced Illumina short reads with PacBio long reads, correspondingly, to produce superior assemblies.
Pipeline modeling of real datasets
Notably, these 105 exons were built using the annotate-my-genome approach, along with one extra exon (referred to as exon 1 below) across SSPO isoforms. These early findings suggest that this technique can address more exons than the methods mentioned, however, this finding may not always indicate a superior assembly. In the simulations, annotate-my-genome combined with a de-novo and/or genome-guided StringTie GTF assembly built from short and long-read alignments beat all mentioned approaches across all examined parameters. Particularly, this pipeline beats all other approaches when combined with genome-guided StringTie assemblies. This latter example supports the tendencies found in the benchmarking using real datasets.
Foolproof checking of the annotate-my-genomes workflow can retain the quality of the StringTie GTF inputs. Evaluation of the F1 scores from the raw and pipeline-processed GTF annotations from –
- de novo StringTie assembly from combined PacBio and Illumina alignments
- assembly of the genome using the pooled PacBio and Illumina alignments, and
- building of a StringTie mix directed by the genome.
The de novo StringTie raw annotations from the Gallus gallus, Mus musculus, Homo sapiens, and Danio rerio datasets were enhanced in actual datasets using the annotate my genomes pipeline at the intron chain, transcript, and locus levels, respectively. Additionally, when StringTie-mix assembly was used as the input for our process, the F1 scores were higher than those obtained from de-novo and merged PacBio+Illumina StringTie assemblies. Significantly, the pipeline tends to retain or even enhance the F1 score attributes from StringTie raw annotations.
In conclusion, it is advantageous to run annotate-my-genome using a genome-guided StringTie GTF file as input when working with hybrid RNA-seq datasets, bringing merged PacBio/Illumina merged alignments or the StringTie-mix approach, given the ubiquity of a good genome assembly including genome annotation in GTF format.
Annotation of crucially missing genes
In conclusion, as opposed to selecting a single annotation tool from either NCBI, Ensembl, or other sources, which is a common practice in next-generation sequencing analysis, our ortholog/paralog assignations of non-annotated coding genes can contribute to the increasing annotation of crucial missing genes and aid in reconciling current annotations. The evolutionary conservation of these processes can also help to uncover new ERVs that may encode functional proteins in vertebrates.
Projected Impact
The scientific community will be significantly impacted by the current study in two ways. On the one hand, this method will make it easier for scientists without sophisticated coding abilities to annotate transcriptome data from hybrid sequencing. This pipeline incorporates industry-recognized techniques for transcriptome annotation and is built as a user-friendly package on the Anaconda/NextFlow/Docker platforms. On the other hand, this study provides an updated annotation that contains an entire transcript with difficult-to-assemble structures, furthering our comprehension of the chicken brain transcriptome.
Implications
This approach is expected to serve biologists looking to enhance transcriptome annotation across a variety of species, tissues, and research domains. Additionally, this dataset will provide a transcriptome resource that the entire community may use to better understand the evolution of certain brain structures. This approach enhances the chicken brain’s existing transcriptome annotation. This pipeline is a convenient system that can be used with a multitude of species, tissues, and research fields to enhance and harmonize existing annotations.
Article Source: Reference Paper | Code & Dataset: annotate_my_genomes
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Riya Vishwakarma is a consulting content writing intern at CBIRT. Currently, she's pursuing a Master's in Biotechnology from Govt. VYT PG Autonomous College, Chhattisgarh. With a steep inclination towards research, she is techno-savvy with a sound interest in content writing and digital handling. She has dedicated three years as a writer and gained experience in literary writing as well as counting many such years ahead.





{kind=link}