
In the dynamic landscape of biology, researchers around the world aim to unlock the secrets held in the genetic code. With the introduction of whole genome sequencing, a new world was opened to science: one where genetic blueprints could be unraveled and studied with ease. Recently, the impact of genome-wide variants on human pathophysiology has become the subject of much investigation. Now, researchers from the University of California, Berkeley, have unveiled a neural network model that promises quick and accurate predictions of the effects of such variants, possessing the potential to change the field of precision medicine.
As whole genome sequencing increases in popularity, much attention has been drawn to the need for a better comprehension of the effects caused by genome-wide variants. If understood, they have the potential to become the basis for new research into precision medicine. Specifically, the ability to predict the damaging effects of a particular variant is important for the diagnosis of rare diseases as well as testing for rare variant burdens.
Language models, made popular by the immense fame of ChatGPT, present a prospective solution to this: trained on large datasets, they are able to predict deleteriousness with great accuracy. Protein language models have been developed that can score missense variants successfully, and these show great promise. However, a similar model hadn’t been developed for DNA so that genome-wide variants can be scored.
Introducing GPN-MSA
For this purpose, the Genomic Pre-trained Network (GPN) was developed: a model utilizing neural networks that were trained on datasets from unaligned genomes, which demonstrated extremely accurate predictions of the damaging effects of certain variants of Arabidopsis thaliana. However, the human genome was significantly more complex than Arabidopsis, containing more repetitive and unfunctional elements and spread out over a much larger area. This makes it much harder to model effects accurately. Prior attempts to garner more accurate outputs resulted in inferior outcomes compared to conventional methods.
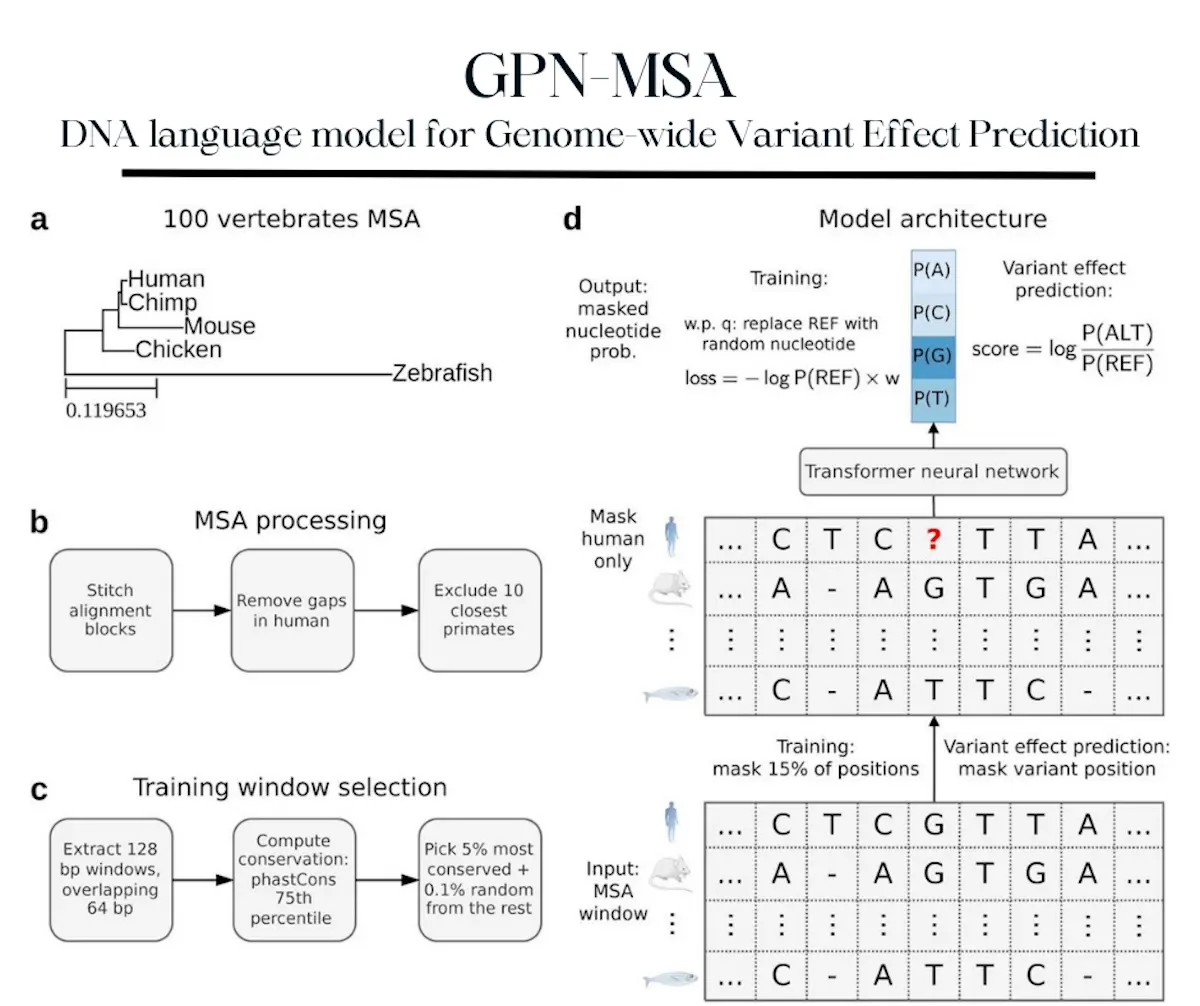
In an attempt to fix this, a new DNA language model has been developed: GPN-MSA. This model has been designed to predict the effects of genome-wide variants based on the incorporation of a multiple sequence alignment of genes from varied species. This was done using Transformer architecture. The model was then applied to humans while using an alignment of 100 vertebrate genomes. It was then shown to outperform past and current DNA language models, some of which are widely used in research today. These include phlyoP, CADD, and Enformer, among others. The model is also much faster than comparable programs, needing only 4.75 hours for training when given 4 GPUs. This is a significant reduction in computing time, making it easier, more efficient, and more user-friendly.
GPN-MSA is an extension of the original GPN, trained to absorb information about nucleotide probability distributions using information from surrounding sequences as well as aligned sequences from other species in order to provide predictions that are significantly more accurate than other similar programs. Inspired by the MSA Transformer, which was trained on multiple sequence alignments from various protein families and was originally developed for the prediction of protein structures before its proficiency at effect prediction was revealed. However, despite these similarities, there are also crucial differences between these models and GPN-MSA, primarily in its training procedures and fundamental architecture. GPN-MSA also works on whole genome DNA alignments, which are considered more complicated than protein sequences due to their varying conservation levels as well as their small and fragmented nature.
Multiple sequence alignments were processed and cleaned, and training windows were selected. Among these, the windows among the top 5% were chosen. Sequences of alignments are then passed through the neural network. The model has been trained so as to upweight conserved elements and downweight repeated elements. Though the reference genome didn’t include the human genome, GPN-MSA was proven to be capable of accurately capturing various attributes of different variants, such as epigenetic modifications as well as the influences of natural selection.
When tested against similar programs for tasks such as classifying missense variants in tumoral cells, GPN-MSA outperforms others in the category by a large margin. The model was then used to classify variants that were known to be linked to the presence of Mendelian diseases. When evaluated on the enrichment of rare variants, the model achieved the highest score overall. It was also used to predict intron variants, where it outperformed SpliceAI, a program developed to give splicing predictions. This success suggests that the incorporation of whole genome multiple sequence alignments is essential and contributes to the accuracy of the results obtained. Variations in the prioritization of conserved regions significantly influence the final results.
Conclusion
The success of this model has important implications for our understanding of the effects of genome-wide variants. Such variants lead to significant changes in the pathophysiology of different individuals, resulting in ineffective treatments. The ability to predict their impact allows for a more comprehensive and holistic understanding of individuals on a case-by-case basis. The utilization of conventional neural networks and language models to provide a rapid, efficient, and accurate method of predicting their effects demonstrates the power of inter-industry collaboration. The use of a language model utilizing whole genome alignments is novel and shows much promise for the assessment and study of the human genome. With the model’s computational efficiency, it is also possible to obtain results rapidly, allowing for research to proceed at a faster pace. Potential applications include genome-wide rare variant burden testing, polygenic risk scores, and functionally informed fine mapping.
Article source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Sonal Keni is a consulting scientific writing intern at CBIRT. She is pursuing a BTech in Biotechnology from the Manipal Institute of Technology. Her academic journey has been driven by a profound fascination for the intricate world of biology, and she is particularly drawn to computational biology and oncology. She also enjoys reading and painting in her free time.











{kind=link}