

The sequencing and assembly of the human Y chromosome have been challenging due to its intricate repeat structure, which encompasses lengthy palindromes, tandem repeats, and segmental duplications. The existing GRCh38 reference sequence lacks over half of the Y chromosome’s content, leaving it as the final unfinished human chromosome. Addressing these limitations, the Telomere-to-Telomere (T2T) consortium introduces the full 62,460,029-base-pair sequence of the human Y chromosome from the HG002 genome (T2T-Y). This new sequence rectifies multiple errors in GRCh38-Y, supplementing the reference with over 30 million base pairs. It reveals complete ampliconic patterns of gene families like TSPY, DAZ, and RBMY, adds 41 protein-coding genes (primarily from TSPY), and unveils a distinctive alternating arrangement of human satellite 1 and 3 blocks within the heterochromatic Yq12 region. Through integration with the CHM13 genome assembly and the inclusion of population variations, clinical variants, and functional genomics data, a comprehensive reference spanning all 24 human chromosomes has been established.
The Y Chromosome: A Tough Nut to Crack in Genetics
The complex architecture of the human Y chromosome, with its large repeats and palindromes, plays a pivotal role in fertility. This includes hosting genes that are crucial for spermatogenesis and sex determination. Although it remains notably incomplete due to over half of its makeup being riddled with gaps in the GRCh38 human reference genome, this condition impedes comprehensive analysis of regions such as Azoospermia factors associated with infertility.
Despite these challenges, breakthroughs from the Telomere-to-Telomere (T2T) consortium have allowed researcher Adam M. Phillippy and his team to assemble the entire CHM13 cell line genome; however, they could not fully put together the Y chromosome because of its unique characteristics. The Human Pangenome Reference Consortium (HPRC) concurrently launched a project to embody a broader genomic range via the use of the HG002 genome. This endeavor successfully resulted in the reconstruction of the full sequence for the Y chromosome, known as T2T-Y.
This achievement paved the way for an extensive analysis of various elements within the Y chromosome. Further integrating T2T-Y with CHM13 assembly gave birth to a novel reference sequence for all human chromosomes—the T2T-CHM13+Y. Through such a monumental resource, enhanced mappability and variant identification becomes feasible, thus promising refined insights into XY individuals’ genetics. These initiatives collaboratively make a significant stride in unraveling the complexities of the human Y chromosome and its implications for genetics and health research.
Assembling and Evaluating the Human Y Chromosome
The assembly and validation of T2T-Y followed a strategic process akin to the T2T-CHM13 genome, employing PacBio HiFi and ONT ultralong reads from the HG002 genome. The initial assembly was executed using a string graph of the entire HG002 genome, with separate components for ChrX and ChrY linked at the pseudoautosomal regions (PARs). Tangles in these XY subgraphs were untangled with ONT reads, and haplotype-specific k-mers from Illumina reads identified chromosomal walks for ChrX and ChrY. Subsequent rounds of polishing involving Illumina, HiFi, and ONT read rectified errors in the draft assembly.
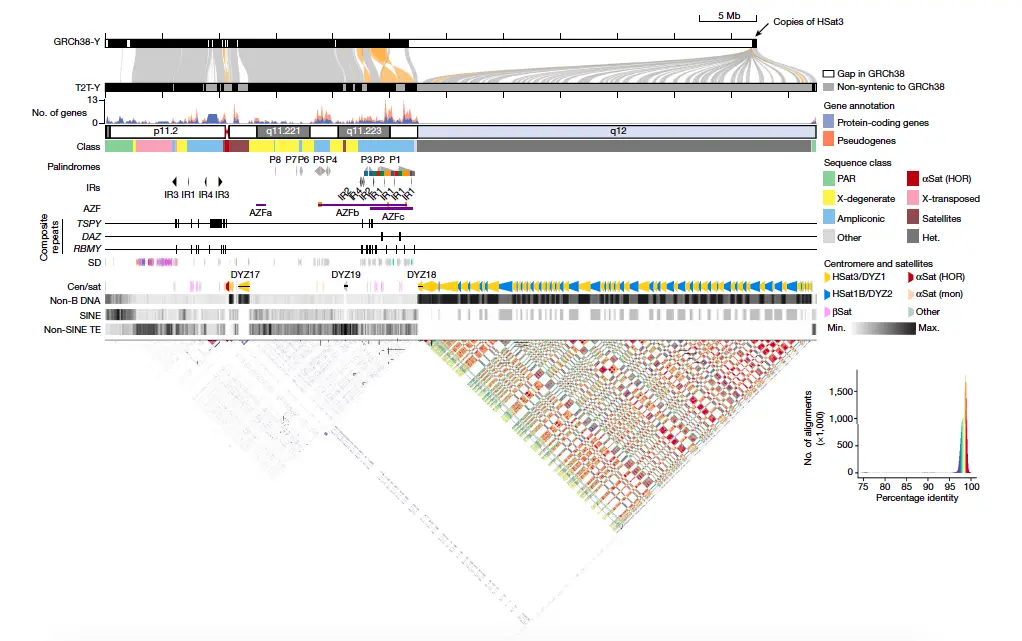
The assembly’s accuracy was verified through various techniques, and the resulting T2T-Y assembly spanned 62,460,029 bases without gaps or model sequences. Notably, this revealed previously uncharacterized sequences within the q-arm heterochromatic region. Compared to the GRCh38-Y reference, T2T-Y exhibited numerous structural differences and an improved sequence identity. The Y-chromosome haplogroups of HG002 and GRCh38 were identified as J-L816 and R-L20, respectively, aligning with their respective ancestries. The combination of T2T-Y with the T2T-CHM13v1.1 assembly produced a new Y-bearing reference, T2T-CHM13v2.0 (T2T-CHM13+Y), completing this significant advancement in genomic research.
Comprehensive Annotation of the Y Chromosome
The T2T-CHM13+Y was comprehensively annotated by incorporating RefSeq and GENCODE annotations from GRCh38 and through manual curation of ampliconic gene arrays. De novo annotations were generated using HG002 full-length cDNA sequencing data, yielding a total of 693 genes and 883 transcripts in T2T-Y. Among these, 106 genes (488 transcripts) were predicted to be protein-coding. Notably, T2T-Y included 110 additional genes compared to GRCh38-Y, with 41 being protein-coding, primarily from the TSPY gene family. The annotation revealed precise information about repeat sequences, including their distribution and variations. T2T-Y was found to contain 8 palindromes, with P1-P3 housing the AZFc region linked to sperm production.
Structural differences and polymorphisms were observed, including a large polymorphic inversion and rearrangements within the heterochromatic region (Yq12). Alpha-satellite arrays with distinct hypomethylated regions and periodic non-B DNA motifs characterized the centromere. Composite repeats associated with genes like TSPY, RBMY, and DAZ were identified, providing insight into their structural organization. The Yq12 heterochromatic region exhibited extensive interspersion of DYZ1 and DYZ2 satellite sequences, with unique structural rearrangements and variations. Phylogenetic analysis shed light on the evolutionary history of these repeats, highlighting connections and seeding events between the HSat1B and HSat3 arrays. These comprehensive annotations contribute to a more precise understanding of the complex genetic architecture of the Y chromosome and its functional elements.
A Novel Approach to Variant Calling
For 3,202 samples from the 1KGP Phase 3 dataset (1,603 XX; 1,599 XY), including 1,233 unrelated XY samples, a comprehensive variant calling approach was conducted. Specifically, 1,233 XY samples were utilized to span diverse 1KGP populations and Y-chromosome haplogroups. The alignment and variant-calling process was enhanced for accuracy and representation by masking ChrY in XX samples and ChrY PARs in XY samples, prompting reads from ChrY PARs to align with ChrX PAR. Diploid genotypes were then inferred within the PAR for both XX and XY samples. Notably, improvements were observed across these XY samples with T2T-Y, including an increased number of mapped reads, a higher proportion of properly paired reads, and fewer mismatched bases compared to GRCh38-Y. The variant counts per sample decreased in syntenic regions of the Y chromosome, particularly pronounced for haplogroup J1. Comparisons between selected samples from different haplogroups revealed the better performance of T2T-Y in terms of variant discovery, detecting fewer variants with excessive read depth and abnormal allele balance. This finding was consistent across a larger sample set from the SGDP. While T2T-Y enhanced short-read alignment and variant calling, a reference alignment was curated between each GRCh reference (GRCh37 and GRCh38) and T2T-CHM13+Y, facilitating the transfer of annotations in both directions. Moreover, most genetic variants from databases like ClinVar, dbSNP, and the GWAS Catalog were successfully lifted to T2T-CHM13+Y, confirming the compatibility and applicability of this improved reference.
Genomic Contamination and T2T-Y
Contamination of genomic databases arises when human DNA fragments are erroneously included in the genomic sequences of other species during assembly. In microbial research, the attempt to filter out such contaminating human DNA using the human reference sequence can be limited due to the reference’s incompleteness. Consequently, portions of human DNA can be mistakenly annotated as bacterial proteins, leading to the presence of thousands of false proteins in public databases. For instance, an analysis of around 5,000 human whole-genome datasets revealed an unexpected association between bacterial species and XY karyotype human samples, with 77,647 100-mers significantly enriched in XY samples.
This phenomenon was hypothesized to result from real human ChrY sequences matched with contaminated bacterial genome entries. Comparing these XY-enriched sequences with the T2T-Y chromosome confirmed this prediction, demonstrating over 95% similarity. Subsequently, the entire NCBI RefSeq bacterial genome database was assessed, revealing 4,179 and 5,148 potentially contaminated sequences aligning with GRCh38-Y and T2T-Y, respectively. These sequences were relatively short and often located in the recently added HSat1B and HSat3 repeats. Such contamination issues are often rooted in repeats due to their high copy number and chances of being sequenced and assembled.
Importantly, this contamination issue likely extends across all human chromosomes and various sequence databases, even beyond microbial genomes. Hence, with improved references like T2T-Y, the contamination problem becomes crucial, as these high-quality references can help identify and rectify such issues in genomic databases.
Conclusion
The comprehensive assembly and annotation of the T2T-Y chromosome represent a significant advancement in genomic research. Through a meticulous process involving PacBio HiFi, ONT ultralong reads, and various sequencing techniques, the T2T-Y assembly was created with improved accuracy and completeness. This assembly, spanning 62,460,029 bases, revealed novel sequences within the heterochromatic region and exhibited structural differences and enhanced sequence identity compared to previous references. The annotation process uncovered a total of 693 genes and 883 transcripts, including additional copies of ampliconic genes such as TSPY. The T2T-Y reference was further utilized for variant calling, revealing enhanced alignment and accuracy across populations and correcting errors present in earlier references. Additionally, the study shed light on the contamination of genomic databases with human DNA sequences, underlining the importance of accurate reference sequences and their impact on genetic research. Altogether, these findings highlight the continuous strides in genomics, providing a valuable resource for understanding the intricacies of the Y chromosome and its implications across diverse research areas.
Article Source: Reference Paper
Learn More:




Neegar is a consulting scientific content writing intern at CBIRT. She's a final-year student pursuing a B.Tech in Biotechnology at Odisha University of Technology and Research. Neegar's enthusiasm is sparked by the dynamic and interdisciplinary aspects of bioinformatics. She possesses a remarkable ability to elucidate intricate concepts using accessible language. Consequently, she aspires to amalgamate her proficiency in bioinformatics with her passion for writing, aiming to convey pioneering breakthroughs and innovations in the field of bioinformatics in a comprehensible manner to a wide audience.











{kind=link}