
The ability to accurately model and redesign ligand binding sites (pockets) on protein surfaces holds tremendous value across fields like drug discovery, enzyme engineering, and biosensing. However, effectively generating protein pockets tailored to recognize user-defined small molecules has remained challenging. Recent research from the University of Science and Technology of China details PocketGen – an integrated deep-learning framework for end-to-end generation of full-atom protein pockets conditioned on target ligands and surrounding protein scaffolds.
The Significance of Protein Pockets
Proteins perform diverse molecular functions by binding to metabolites, toxins, hormones, and pharmaceutical agents through specific pockets on their surfaces. These pockets comprise intricate 3D microenvironments facilitating selective, high-affinity interactions with collaborating molecules.
Such temporary associations can profoundly influence overall protein behavior. For example, binding events often trigger structural rearrangements that modulate or inhibit activity. Therefore, pockets act as vital regulatory handles, enabling external control over protein functions.
Designing custom pockets presents opportunities to rationally augment proteins with desired molecular recognition capabilities. Specific applications include:
- Directed enzyme evolution to transform metabolic pathway fluxes
- Biosensor/assay development for detecting toxins or biomarkers
- Therapeutic antibody maturation to improve treatment potency
However, reliable computational design of tailored pockets has remained extremely difficult thus far. The process necessitates coordinated optimization of 3D geometry and amino acid sequences to mold complementary interfaces – all while accounting for molecular flexibility.
Generalized tools capable of directly generating application-specific binding sites could massively expand the functional repertoire of proteins. However, modeling interdependent, multi-scale biochemical relationships simultaneously has continued to pose barriers.
Prior Computational Approaches
A variety of computational strategies have been historically attempted for pocket design with limited success:
- Physics-based simulations tune predicted energies between atoms and search for optimized pockets using biophysical principles. However, conformational exploration and binding affinity forecasts employing brute-force physics incur massive computational expenses.
- Template-matching methods transfer pocket structural motifs from previously characterized protein-ligand complexes to design binding sites. However, the availability of well-matched templates across diverse ligands is inconsistent at best. Conformational strains and distortions frequently arise as well from grafting.
- Earlier deep generative models like RFDiffusion first generate protein backbones before predicting sequences. But such decoupled tactics produce incompatible pockets with poor affinity owing to steric clashes.
In essence, simultaneously balancing flexibility, sequence dependencies, and microscopic interactions has continued to impede both physics-driven and data-driven approaches thus far.
Introducing PocketGen
To address the preceding challenges, PocketGen implements an integrated deep-learning architecture with the following differentiating capabilities:
- Joint sequence and structure refinement using a co-design scheme
- Multi-scale interaction modeling via graph networks
- Incorporation of evolutionary constraints through protein language models
- Structural cross-attention to enforce sequence-structure consistency
This combination enables end-to-end generation of full-atom binding site structures conditioned on target ligands and surrounding protein environments. The choreographed components work synergistically to produce physically plausible pockets exhibiting designed-in specificity.
Key Innovations Under the Hood
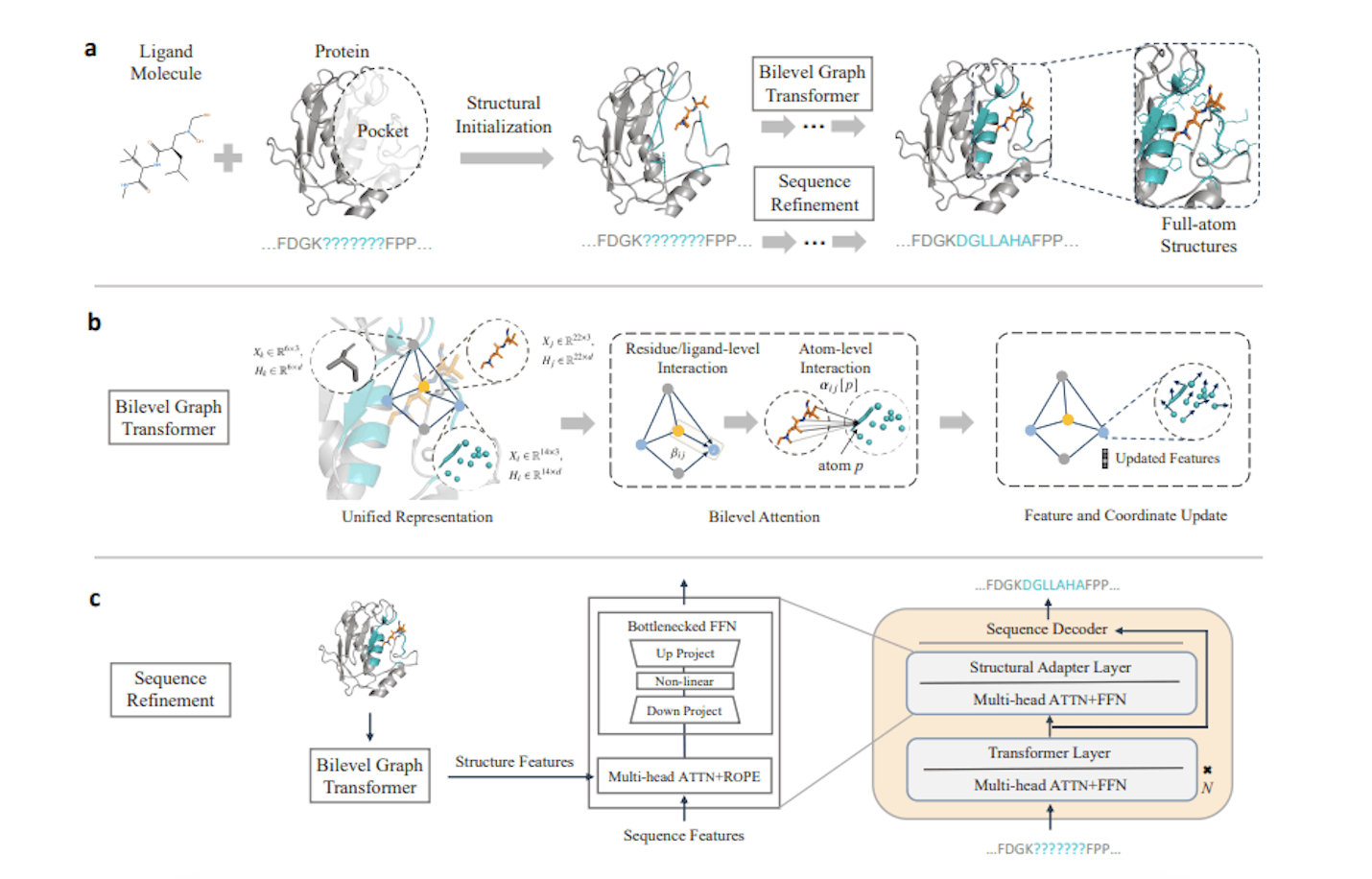
PocketGen comprises two modules working in unison: a multi-scale graphical transformer fusing coordinates and protein language models bearing evolutionary knowledge.
Bilevel Graph Transformer
This component leverages graph networks to model inter-residue relationships within the binding site at two integrated scales:
Atoms: Capture fine-grained non-covalent interactions critical for molecular recognition specificity.
Residues & Ligands: Provide coarser representations that are less sensitive to inconsequential perturbations.
This representation also remains unchanged irrespective of rotations/translations (a useful equivariance property).
Multi-grain information then flows bidirectionally through bilevel attention layers to recursively update node embeddings and pocket coordinates – thereby reflecting interdependent pocket-ligand relationships.
Sequence Refinement with Protein Language Models
Separately, a protein language model infuses the design process with rich evolutionary constraints and long-range dependencies lacking in purely structural representations.
To harmonize these distinct knowledge spheres, the authors implant a lightweight adaptation module permitting controlled transfer learning. Cross-attention mechanisms allow balanced gradient flows while minimizing added parameters through freezing.
This strategy enforces sequence-structure consistency during pocket generation through computationally efficient interface learning.
Training Methodology
The training data comprised CrossDocked and Binding MOAD datasets of protein-ligand complexes. Only the adapter modules reconciling structure and sequence pathways were tuned through backpropagation. By keeping language model foundations frozen, available evolutionary knowledge was transferred without disruption.
The composite loss function enforced penalties on coordinate deviations, bond lengths, residue types, and ligand poses simultaneously. Additional regularizers were introduced programmatically to improve generalizability as well.
PocketGen outperformed predecessor protein generation techniques on various standardized benchmarks – confirming reliable convergence.
Evaluating Model Performance
Design Capabilities on Standard Benchmarks
Quantitative benchmarking reveals PocketGen’s consistent ability to produce valid pockets exhibiting:
- Higher predicted binding affinities
- Improved backbone geometries
- Greater sequence recovery rates
- Enhanced structure-sequence compatibility
These comprehensive gains demonstrate the value of interdependent sequence-structure co-design under this integrated framework.
Demonstrated Utility on Pharmaceutical Targets
Critically, when tasked with redesigning binding sites on therapeutically relevant proteins, PocketGen generated models displaying plausible interactions, improved predicted stability, and increased shape complementarity with drug molecules.
The cases spanned antibody maturation for sensors, coagulation factor remodeling for anticoagulants, and opioid-binding proteins for abuse detection – confirming expansive applicability.
Diagnostic Analyses
By visualizing model attention weights as well, the researchers confirmed PocketGen’s capacity to implicitly derive meaningful biochemical relationships driving inter-residue contacts, hydrogen bonds, and aromatic stacking interactions – further underscoring interpretability.
Conclusions & Future Outlook
PocketGen’s pioneering methodology sets new standards for designing custom pockets to confer desired ligand specificities in proteins – substantiated through well-rounded benchmarking. The grounded architectures, real-world utility spanning drug discovery contexts, and interpretability collectively showcase a powerful, extensible platform.
Looking ahead, active research pursuits involve augmenting model robustness on less characterized protein families, as well as improving computational efficiency to enhance accessibility for a variety of bioengineering groups.
Downstream wet lab testing on generated pockets should also lend vital empirical validation toward future translational efforts. Accessible automated tools capable of reliably designing pockets have the potential to accelerate candidate screening and de-risk experimental pipelines pursuing enzyme engineering or precisely targeted therapeutics.
Indeed, as personalized medicines and sustainable biocatalysts continue garnering attention, AI-assisted binding site remodeling techniques like PocketGen may soon grow into indispensable solutions supporting various precision bioengineering programs.
While long-term real-world impact awaits further innovation, the initial strides reported here demonstrate how synergistically merging modern statistical and classical biochemical insights promises increasingly agile yet accurate pocket modeling capabilities – providing reasons for optimism as researchers march forward.
Story source: Reference Paper | The source code of this study is freely available on GitHub
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:



Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}