
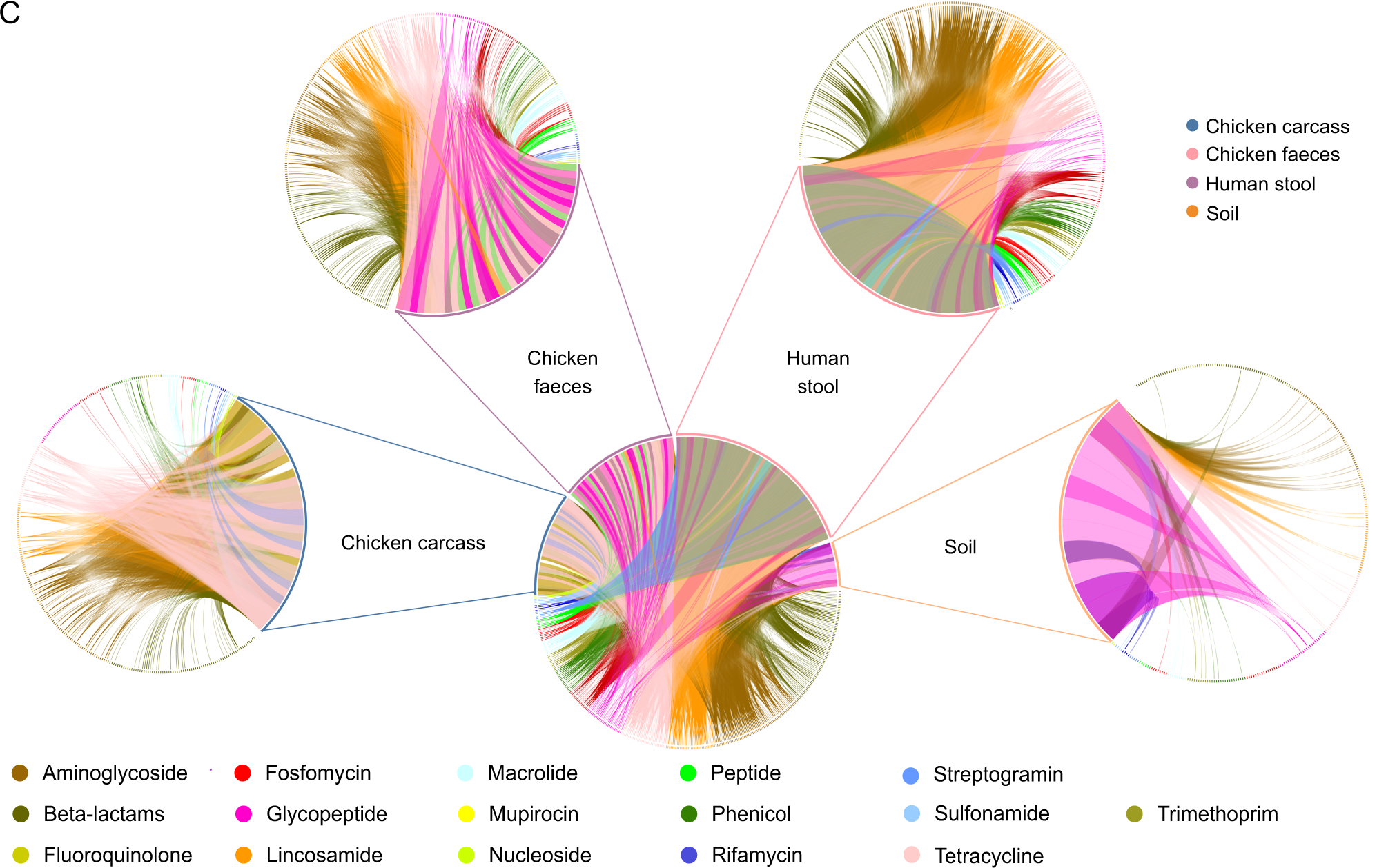
Antimicrobial resistance (AMR) may be reservoired in intensive livestock farms, posing a threat to surrounding communities. The gut microbiome of livestock, workers, and their households, as well as microbial communities in carcasses and soil, were investigated using shotgun metagenome sequencing, machine learning (ML), and culture-based methods at a poultry farm in China. In this study, both microbiomes and resistomes differed across environments and hosts. Both human and broiler chicken samples showed several clinically relevant antimicrobial resistance genes (ARGs) and related mobile genetic elements. AMR surveillance on farms requires close monitoring of Escherichia coli. It was found that strains of E. coli were present in both humans and chickens. In chicken fecal resistomes, several ARGs correlated with the resistance/susceptibility profiles were isolated from E. coli culture from the same samples. Environmental sensing has additionally revealed that variation in temperature and humidity has a significant impact on these ARGs. In complex, interconnected environments, it is important to adopt a multi-domain and multi-scale approach when studying microbial communities and AMR.
Image Source: https://doi.org/10.1038/s41396-022-01315-7
There is a major global concern regarding antimicrobial resistance (AMR), and surveillance of AMR has historically focused on clinical settings and high-income countries (HICs). In many low- and middle-income countries (LMICs), resistant bacteria are able to circulate largely undetected outside of clinical settings. Since livestock and humans may have more contact in LMICs, antibiotics are used for growth promotion and metaphylaxis at a higher rate than in HICs. Eggs and meat from poultry are major sources of animal protein for human consumption.
During the past 60 years, chicken meat production has increased by more than 1500%. Due to increasing demand for animal protein, some countries, particularly LMICs, have shifted towards livestock production systems that rely on antimicrobials to maintain animal health and productivity. During the past three decades, industrial livestock production has expanded in China, where this study was conducted. In 2030, China will use 30% more antibiotics than any other country (23%) in animal production.
Animals’ guts have been shown to be altered and expanded by antibiotic usage, even at low levels. All microorganisms lining the gut contain antibiotic resistance genes (ARGs), including bacteria, viruses, archaea, and eukaryotes. ARGs have been found to be transmitted between livestock and humans via the gut microbiota, despite the microbiota being largely commensal and important for health.
It has been found that antibiotic-resistant bacteria (ARBs) and antimicrobial resistance genes (ARGs) are present both directly (in food production) and indirectly (through food consumption). Some studies have shown differences in human and animal lineages when comparing samples from different regions, indicating that farm animals may play a significant role in developing and disseminating ARBs.
There is a great deal of complexity and constant change in the gut resistome. By identifying which ARGs pose the greatest threat to public health, it may be possible to identify the species that pose a greater threat by analyzing how such variability is related to resistance phenotypes in the commensal bacterial community of the gut and of the pathogens in the gut. In the same way, by investigating markers associated with resistance phenotypes in indicator bacteria, we can begin to identify additional risk factors for hotspots of resistance other than those directly related to those markers. The exploration of these research gaps may further shed light on the complex relationship between AMR phenotypic presentation and the reservoir of ARGs within the resistome where the pathogen lives. In order to examine correlations between phenotypic resistance and resistome, culture-dependent and culture-independent methods can be combined. ML is ideally suited for studying AMR in this way because it is efficient at analyzing datasets with complex variables.
The metagenomic data has recently been combined with culture-based analyses. However, none of these studies have correlated resistance phenotypes to metagenomic data. AMR and virulence determinants of clinical infections have been predicted using metagenomic samples (by comparing known genes in public databases); however, in typical studies, these samples are monomicrobial or polymicrobial with only two species present. AMR has already been well demonstrated in the wider microbiome context by bacterial species like Escherichia coli. It may be possible to predict species, genes, and MGEs correlated to AMR by linking metagenomic data to bacterial AMR phenotypes taken from the same samples. To date, no ML studies have utilized metagenomic data in this way, and this study attempts to fill this gap.
Further, external factors, such as temperature or humidity, may also influence phenotypic changes when linking phenotypic changes to the resistome. Microorganisms in the gut are normally stable, but when perturbing factors intervene (e.g., dietary changes, infections, stress, antibiotic administration, environmental changes), they change. Changing bacteria may transfer resistance to commensals or result in the establishment of new resistant bacteria. Despite the need for mesocosm studies to shed light on factors influencing the resistome, little information about specific factors associated with resistome changes is available, and systemic approaches should integrate such information for a deeper understanding.
As performance indicators such as sensitivity, specificity, and reproducibility of the methods need to be improved, ML protocols and bioinformatic tools for evaluating the association of the resistome and resistance phenotypes are not readily available. Additionally, these methods are not feasible for implementation globally, especially in LMICs. A larger sample size and a broader geographical area should be considered before this study can be deemed proof of principle. If the method is validated at larger spatial, geographical, and temporal scales, it might provide novel solutions for epidemiological monitoring suitable for use in low- and middle-income countries. Especially when considering how this knowledge and methods may be applied to mitigate AMR within the context of increasing adoption of sensing and monitoring technologies, digitalization in livestock farming, and the implementation of precision poultry farming with the latest ML and big data mining technologies.
Article Source: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Srishti Sharma is a consulting Scientific Content Writing Intern at CBIRT. She's currently pursuing M. Tech in Biotechnology from Jaypee Institute of Information Technology. Aspiring researcher, passionate and curious about exploring new scientific methods and scientific writing.





 may be reservoired in intensive livestock farms, posing a threat to surrounding communities.){kind=link}