
A new powerful machine learning tool, ‘SigProfilerExtractor‘ has identified a link between bladder cancer and tobacco smoking. The study was led by researchers at the University of California San Diego School of Medicine and published in the journal Cell Genomics. The study may help researchers determine what environmental factors could cause cancer in certain patients, such as exposure to tobacco smoke and UV rays.
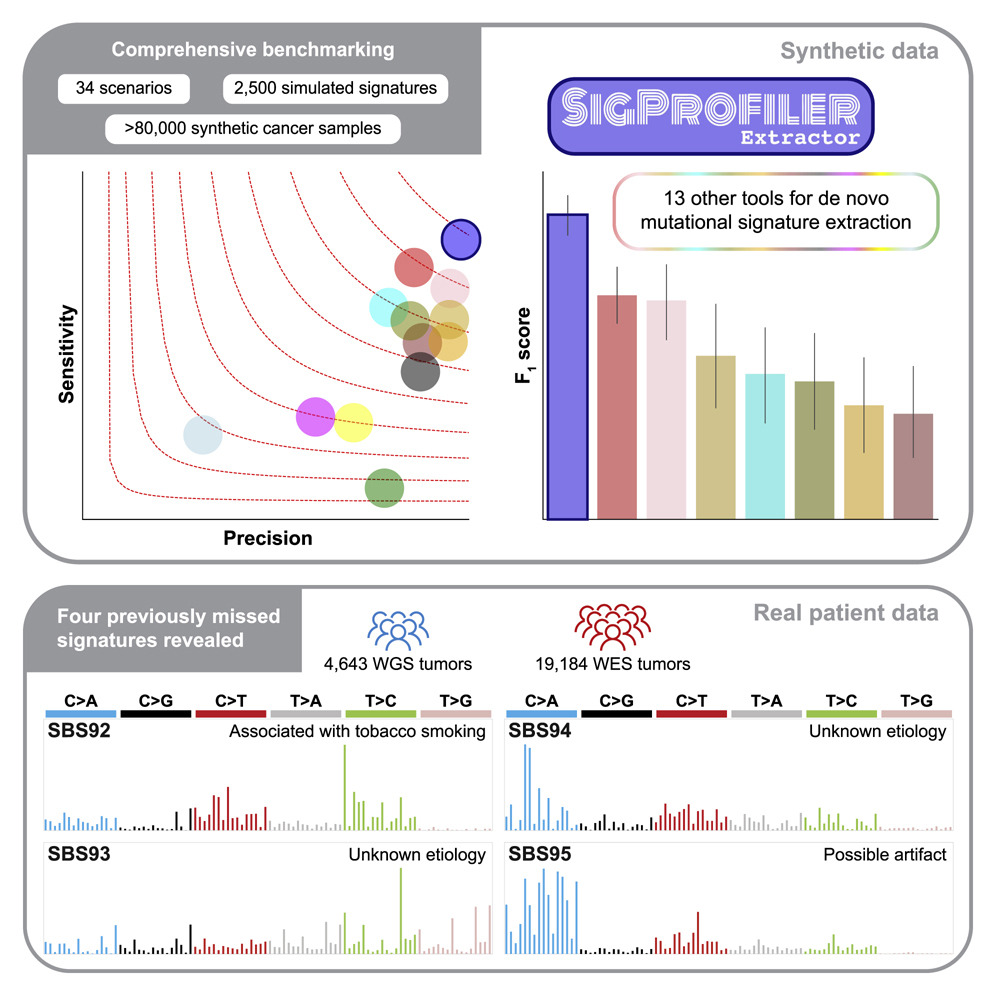
The analysis of mutational signatures is commonly used in cancer genomic studies. This study uses 34 scenarios encompassing 2,500 simulated signatures found in 60,000 synthetic genomes and 20,000 synthetic exomes to benchmark SigProfilerExtractor against another 13 bioinformatics tools and to present SigProfilerExtractor, an automated tool for de novo mutational signature extraction.

SigProfilerExtractor outperforms other approaches in simulations with 5% noise, reflecting high-quality datasets by revealing 20% to 50% more true-positive signatures while yielding five times fewer false positives. Four novel signatures can be identified by applying SigProfilerExtractor to whole-genome and whole-exome sequences of 4,643 cancers. One of the signatures is associated with tobacco smoking, and two are confirmed in independent cohorts. A comprehensive benchmarking of bioinformatics tools for extracting mutational signatures is presented in this report, as well as several novel mutational signatures, including one attributed to direct tobacco smoking mutagenesis in bladder tissues.
Cancer genomes contain somatic mutations that result from endogenous and exogenous mutational processes that have occurred throughout the cell’s lineage. A typical pattern of somatic mutations has been identified for specific environmental carcinogens by examining TP53 mutations in cancers. Next-generation sequencing data and novel computational approaches have enabled the separation of the signatures of individual mutagenic processes in cancer genomes. There are more than 100 distinct signatures in cancer genomes, some of which are related to environmental carcinogen exposure, failures of DNA-repair pathways, infidelity or deficiency of replicating polymerases, and iatrogenic events. Both cancer prevention and cancer treatment have been aided by mutational signatures.
Existing tools for de novo extraction of mutational signatures:
- Support the SBS-96 mutation classification system, which encompasses single base substitutions across 50 and 30 sequences.
- Fail to determine the number of signatures automatically.
- Failed to identify a robust solution.
- Require preselection of many hyperparameters.
- Do not decompose de novo signatures into over 100 reference signatures available in the Catalog of Somatic Mutations in Cancer (COSMIC) database.
De novo extraction tools have not been extensively benchmarked, which leaves uncertainty about their performance.
The purpose of this paper is to address these limitations by providing SigProfiler Extractor-a reference tool for the extraction of mutational signatures de novo. It analyzes all types of mutational classifications, performs an automatic selection of signatures, yields robust solutions, requires only minimal setup, and decomposes de novo extracted signatures to known COSMIC signatures. With 3,608 unique matrix decompositions across 34 distinct scenarios using SigProfilerExtractor and 13 other tools, a comprehensive benchmark demonstrated that SigProfilerExtractor can handle noise effectively and outperforms all other computational tools in the extraction of mutational signatures de novo.
The PanCancer Analysis of Whole Genomes (PCAWG) project recently published 2,778 whole-genome-sequenced (WGS) cancers, along with 1,865 whole-genome sequencing (WGS) and 19,184 whole-exome sequencing (WES) cancers, revealed four novel mutational signatures using SigProfilerExtractor. The SBS92 signature is associated with tobacco-associated mutagenesis and has been confirmed in two independent cohorts.
For de novo extraction of mutational signatures from noiseless datasets as well as from datasets containing various levels of random noise, including synthetic data emulating WGS and WES cancers, SigProfilerExtractor outperforms 13 other tools. When compared to any of the other tools, SigProfilerExtractor generates fewer FP signatures and identifies more TP signatures. Using a model-selection algorithm, de novo extraction determines the number of operative signatures based on both a factorization approach and a model-selection algorithm.
With the forced model selection, SigProfilerExtractor’s factorization performs better than that of other tools when extracting the known number of ground-truth signatures. Further demonstrating SigProfilerExtractor’s ability to uncover novel biological results is benchmarking with suggested model selection, which matches the analysis of a real dataset most closely. The analysis of these downsampled genomes by SigProfilerExtractor failed to detect SBS92, confirming that exome sequencing alone is not sufficient for identifying the signature SBS92.
SigProfilerExtractor is a computational tool for de novo extraction of mutational signatures. The largest benchmarking of bioinformatics approaches for extracting mutational signatures demonstrates that SigProfilerExtractor outperforms 13 other tools. A further application of SigProfilerExtractor revealed four mutational signatures in bladder cancer and in normal bladder epithelium, including one associated with tobacco smoking mutagenesis.
Optimizing the tool to make it more user-friendly and personalized
Across the genomic landscape, mutational signatures are assumed to accumulate linearly and independently. The assumption may hold true for most signatures of small mutational events, such as substitutions, insertions, and deletions, but it may be challenged by larger mutational events, such as most copy-number signatures. Moreover, a prior study demonstrated that at least one substitution signature is not a superposition of individual alterations. Due to their frequent occurrence in cancers with concurrent loss of both polymerase proofreading and mismatch repair, current benchmarking ignores such scenarios. Last but not least, the study focused on benchmarking the de novo extraction of mutational signatures from large sets of tumor samples, not on assigning signatures to particular cancer genes. Future benchmarking efforts will be necessary to determine the accuracy of the assignment of mutational signatures to specific cancers.
The team hopes to develop a web-based tool that can be used by more researchers to profile more patients.
Article Source: Reference Paper | Codes and Data Availability: GitHub | Installation, Usage, Explanation: Wiki Page
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Srishti Sharma is a consulting Scientific Content Writing Intern at CBIRT. She's currently pursuing M. Tech in Biotechnology from Jaypee Institute of Information Technology. Aspiring researcher, passionate and curious about exploring new scientific methods and scientific writing.





{kind=link}