
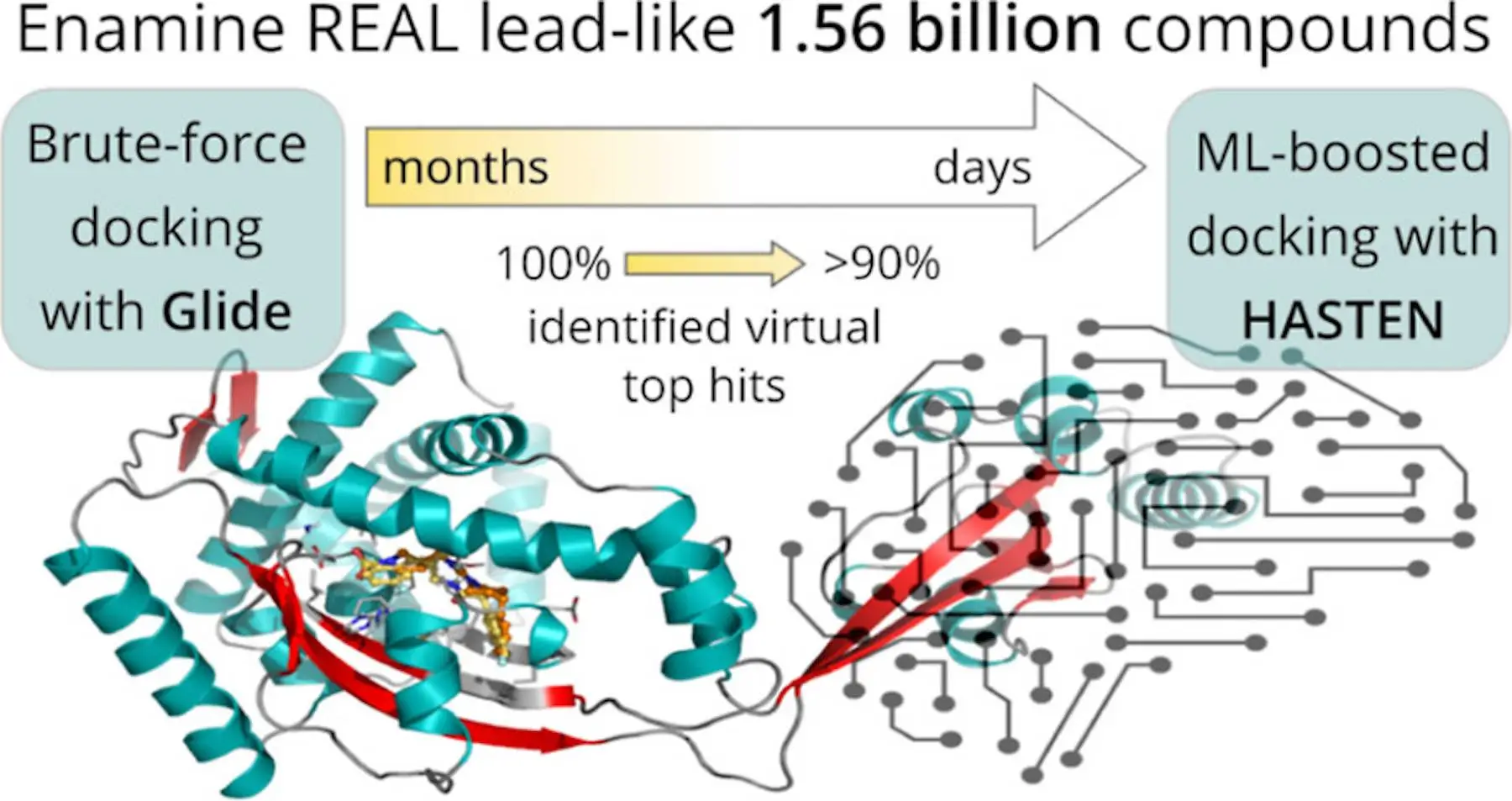
A team of researchers utilized HASTEN, a machine-learning (ML) model, to perform virtual screening on ultra-large chemical compound libraries. Several challenges are faced when screening libraries of such large scale, and conventional brute-force docking methods that are usually used have drawbacks, especially for libraries that span nearly a billion compounds. HASTEN is a quick and efficient method to boost docking methods, reducing the time period and computational power consumed while performing such screenings. It has been used to study an antiviral kinase and an antibacterial chaperone as primary targets and has contributed to benchmarking virtual screening (VS) procedures.
The role of molecular docking in early-stage drug discovery
In the early stages of drug discovery projects based on the structures of compounds, virtual screening of chemical libraries is commonly carried out using molecular docking. It is a method used to predict the interactions between proteins and ligands; ligands are also referred to as ‘small molecules’. The main goal of docking is to determine the small molecular ‘hits’ that are most likely to bind with unknown target proteins. ‘Hits’ is a term used to refer to the identification of small molecules that have successfully interacted with the target protein.
Large libraries that contain compounds already validated by biochemical assays are preferred for screening to avoid spending additional time performing organic synthesis of the compounds. These libraries have continued to grow over time and, as of now, have managed to accumulate billions of compounds. While this does increase the diversity of the dataset used for docking, it makes conventional docking unfeasible in the long run due to the immense amount of time it would take.
Tackling the challenges that come with screening giga-scale libraries
This study takes a structure-based approach to virtual screening, which in turn yields better-quality results with higher resolution than the conventional docking methods that were mentioned previously. This study was used to analyze two targets: the first was the D4 dopamine receptor, and the second was the β-lactamase AmpC. They were docked to nearly 100 million compounds, and the researchers ended up discovering new ligands for the targets as well.
Several approaches have been hypothesized, tried, and tested to deal with the computational expenses that come with screening libraries that span billions of chemical compounds. One of them proposes that instead of docking molecules in their full size and form, it would be better to grow new compounds using their fragments. This reduces the number of compounds that would be required for docking.
With recent technological developments that have taken place over the past few years, ML methods have started to become the method of choice for performing virtual screening for researchers across the globe. The basic principle used here is simple: compounds that have been conventionally docked beforehand are used as training datasets for ML models.
The first suggested ML approach for virtual screening describes the classification of hits into ‘virtual hits’ and ‘non hits’; an example of this is DeepDocking. The second approach suggests predicting docking scores for the entirety of the library, eliminating the need for docking each and every compound. This approach uses a combination of HASTEN and Glide Active Learning.
What is HASTEN?
HASTEN is a software developed to expedite the process of structure-based virtual screening in drug discovery. HASTEN’s abbreviation is derived from macHine leArning booSTEd dockiNg. Chemprop, an ML ‘engine’, is used for iteratively predicting docking scores for screened compounds. Training data from previously docked compounds is used for predicting scores as well as the compounds’ SMILES representations. To validate its efficacy, datasets containing millions of molecules that have been previously docked by FRED were used. For the top 1% docked molecules scored by HASTEN, a recall value of 0.78 was noted, showing decent performance by the ML model. For this study, molecules that were docked in-house by Glide were used, which ended up exhibiting an excellent recall value of 0.95.
A deep dive into the model
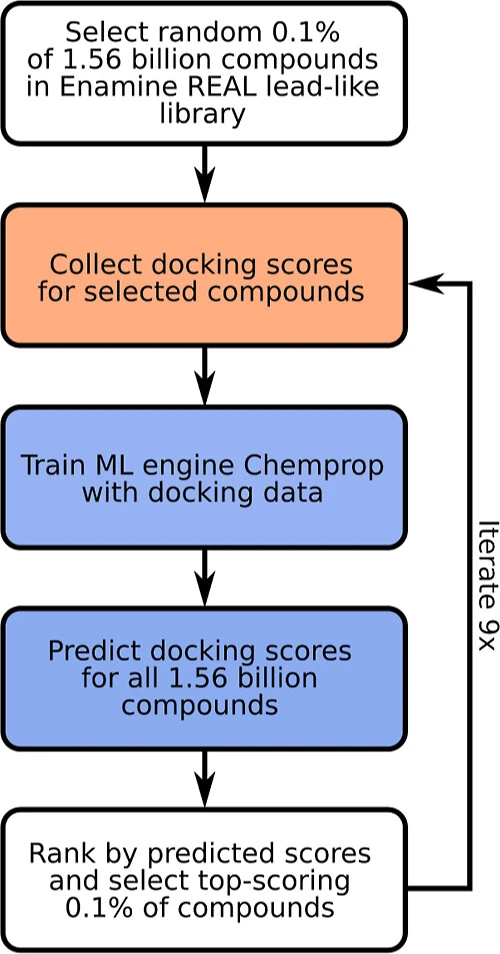
Image Source: https://doi.org/10.1021/acs.jcim.3c01239
Machine Learning performance is enhanced by using regression models such as random forests (RF) or a combination of RF and graph convolutional neural networks (GCNNs), such as Glide Active Learning.
The compounds are ranked according to their scores. The top rankers are selected for the next iterative round; this step exponentially improves the recall abilities of the model. Recall is a metric used to assess the ability of the docking method to identify binding regions accurately.
Glide high-throughput virtual screening (HTVS) was used to dock compounds from the same library and set of compounds used as HASTEN’s training datasets. This made the study more comparable. The researchers also happened to carry out one of the largest conventional docking campaigns ever held in the past few years.
Two targets were used for the ML model’s training dataset and were studied extensively:
- SurA protein: It is a periplasmic chaperone that resides in Gram-negative bacteria. It transports outer membrane proteins and is involved in their maturation as well. It is a target for understanding and tackling antibiotic resistance exhibited by bacteria, as this chaperone contributes to reducing their resistance and makes them more sensitive to antibiotics.
- Cyclin G-associated kinase (GAK): It is a serine/threonine kinase. It regulates the trafficking of clathrin, a vesicle-coating protein, and its role in the mediation of endocytosis. It is an important antiviral target, as it acts as a host factor in regulating the entry of viruses such as Ebola and hepatitis C into the human body.
The library screened in this study was the Enamine REAL lead-like screening library (ERLL). Docking data is rarely available for public use or viewing. The researchers benchmarked and released the docking datasets utilized in this study to the public.
The model is capable of dropping failed compounds, thereby reducing the number of mislabeled compounds. This makes the scoring process easier and more robust and aids the machine-learning model in easily associating unknown compounds with their respective scores.
Conclusion
The boosted docking model’s combination of HASTEN and Glide SP displayed a much larger fraction of docking (0.1%) compared to the much smaller fraction (0.01%) of the HTVS-docking results.
This boosted model has the potential to save more time by using strategies such as earlier stopping, only leading to a slight decrease in the recall of virtual hits. Hyperparameter optimization is another option that can be considered to improve the recall abilities and the model’s speed.
The HASTEN-boosted approach for docking compounds to screen extremely large libraries is a very efficient approach that has reduced the calculations required for docking by nearly 99%. It has the potential to tackle any future challenges posed by the ever-increasing volumes of compounds gathered every year in chemical compound libraries and can accelerate drug discovery research and development.
Article source: Reference Paper | The tool HASTEN is freely available on GitHub
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.











{kind=link}