
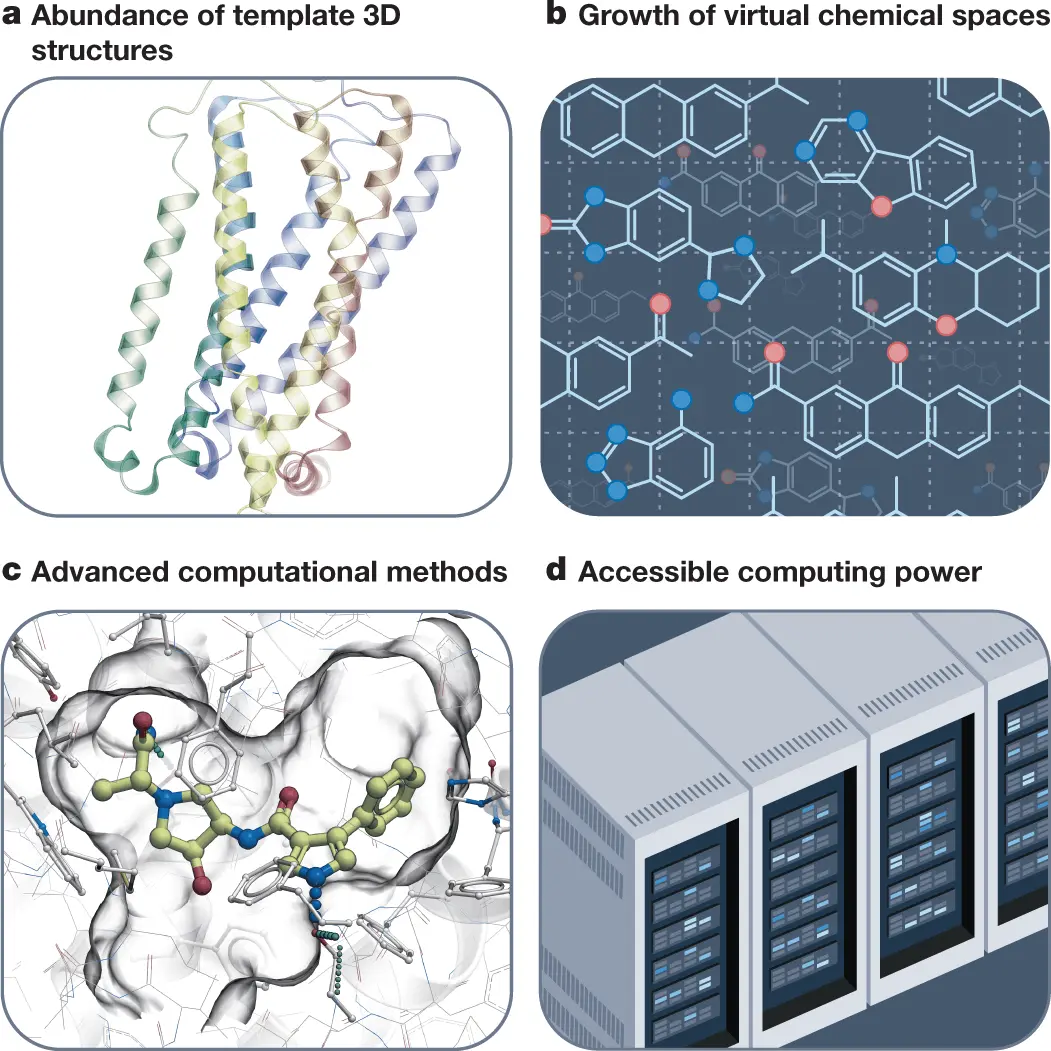
Scientists from the University of Southern California, USA, shed light on the computational approaches that are transforming the drug discovery process from computer-aided to computer-driven. The flood of data about ligand properties and binding to therapeutic targets, coupled with their 3D structures, has revolutionized the drug discovery methodology. The advancement of computing capacities and the advent of on-demand virtual libraries comprising numerous drug-like small molecules have further added to the computer-driven drug discovery process. Ligand screening, aided by fast iterative screening technologies and virtual screening of gigascale chemical spaces, has generated enormous amounts of potential drug candidates. Deep learning-based approaches for predicting ligand properties are an added advantage. The authors envision that computer-driven drug discovery will lead to cost-effective development for better therapeutics.
Computer-aided drug discovery to computer-driven drug discovery
Computer-aided drug discovery was formalized in the early 1970s and became popular with Fortune magazine in the early 1980s. Since then, there have been cycles of hype as well as disillusionment with computer-aided drug discovery. Computer-aided approaches have become an integral yet modest part of the current drug discovery process, and needless to say, there have been several success stories.
The recent advancement in technology, coupled with several scientific breakthroughs, have rendered the computational approaches the key driving paradigm for the Drug Discovery procedure. Both in academia as well as in the industry, the significance of computer-driven drug discovery is noteworthy. Several biotech companies have raised billions by incorporating business models based on molecular modeling using advanced physics, deep learning, and artificial intelligence paradigms. While expecting novel approved drugs solely out of a computational pipeline without the need for experiments is still far from reality, the vast number of potential candidates being generated is remarkable indeed. In fact, some of the campaigns are promising a target-to-lead time of as less as 1-2 months. Only time will tell if this is another cycle of hype or disillusionment, but what cannot be disregarded is the streamlining of the drug discovery process in a computer-driven manner.
Key factors driving the change
The structural revolution, the rapid expansion of drug-like chemical space, and the emergence of computational approaches incorporating 3D structures and ligand data are the main factors driving the transformation of the drug discovery process.
Structural biology has evolved over the years in leaps and bounds, from automated crystallography to microcrystallography, as well as the cryo-electron microscopy technology that has revealed 3D structures for several clinically-relevant targets. The new addition, AlphaFold, has indeed further expanded the landscape of available 3D structures.
The rapid expansion of drug-like chemical spaces has increased the possibility of discovering potential candidates. While this number was limited to several millions of on-shelf compounds, with rapid expansion, it has exploded to billions. Screening procedures are now aided by ultra-large virtual libraries.
The availability of high-performance computing facilities such as cloud computing and GPU-based computing has greatly added to the streamlining of the drug discovery process. With the availability of large pools of ligand data and easily accessible 3D structures, such computing capabilities have remarkably transformed the drug discovery methodology.
The authors elaborately review the ongoing expansion of the drug-like chemical spaces as well as the computational approaches for ligand discovery and optimization.
The bigger, the better: expansion of accessible chemical space
One of the critical bottlenecks in the drug discovery process has been the limited number of screening libraries for potent ligand detection. An affordable high-throughput screening (HTS) procedure makes use of approximately 50,000 to 5,00,000 compounds, yielding only very few hits. These hits often have suboptimal pharmaceutical properties necessary for potential drug candidates. On the other hand, virtual libraries generated using in silico techniques have resulted in a massive expansion of the screening libraries, thereby increasing the chances of finding potential drug candidates. The chances of finding ligands with optimal properties are also greatly improved in this vast ligand landscape. The REAL database was the first commercially available on-demand library. SAVI ( synthetically accessible virtual inventory) was developed by the NIH, USA.
Computational approaches driving the transformation
With the expanded ligand space and the ever-increasing virtual libraries for screening, the availability of highly advanced computational approaches is a necessity. Gigascale screening requires high-performance computing facilities, as well as caution is necessary for safeguarding against the generation of false positives at such large scales. Ligand scoring functions are devised to tackle this challenge. They are designed to remove non-binders, thereby minimizing false positives.
Modular synthon-based approaches have resolved the task of designing molecules from a few fragments that optimally fill up the receptor binding pocket. LUDI and V-SYNTHES are such algorithms.
Data-driven and deep learning-based approaches have truly revolutionized the drug discovery process. ML paradigms such as Support Vector Machines, Random Forests, and neural networks have been used for ligand property prediction. With the advent of the deep learning revolution, drug discovery has also benefited from it in leaps and bounds. The learning-from-examples paradigm of the deep learning-based approach is highly useful in extracting general models of binding affinities.
Conclusion
Drug discovery and development have vastly benefited from the enormous advancement in computer-aided drug design. The advent of technological advances in terms of computation capabilities, as well as the application of artificial intelligence and deep learning paradigms, have remarkably reshaped the drug discovery procedure and have rendered it heavily dependent on computational approaches. What was once computer-aided is now being driven by computational approaches, and the authors envision that in the future, the discovery of novel drugs will no longer be time-consuming and tedious but rather fast-paced, generating better therapeutics.
Article Source: Reference Paper
Learn More:




Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}