
Scientists from the Center for the Study of Systems Biology, Atlanta, in collaboration with the Georgia Institute of Technology, Atlanta, and Oak Ridge National Laboratory, Oak Ridge, USA, have introduced a new method, ‘AF2complex’, in which AlphaFold2 (AF2) can be used to predict protein complex structure and protein-protein interactions. It is expected to provide significant structural insights into various biological molecular systems.
The structure of protein complexes can be predicted at much higher accuracy than classical docking approaches by adapting AlphaFold2, even if the docking methods use monomeric structures predicted by AlphaFold2.
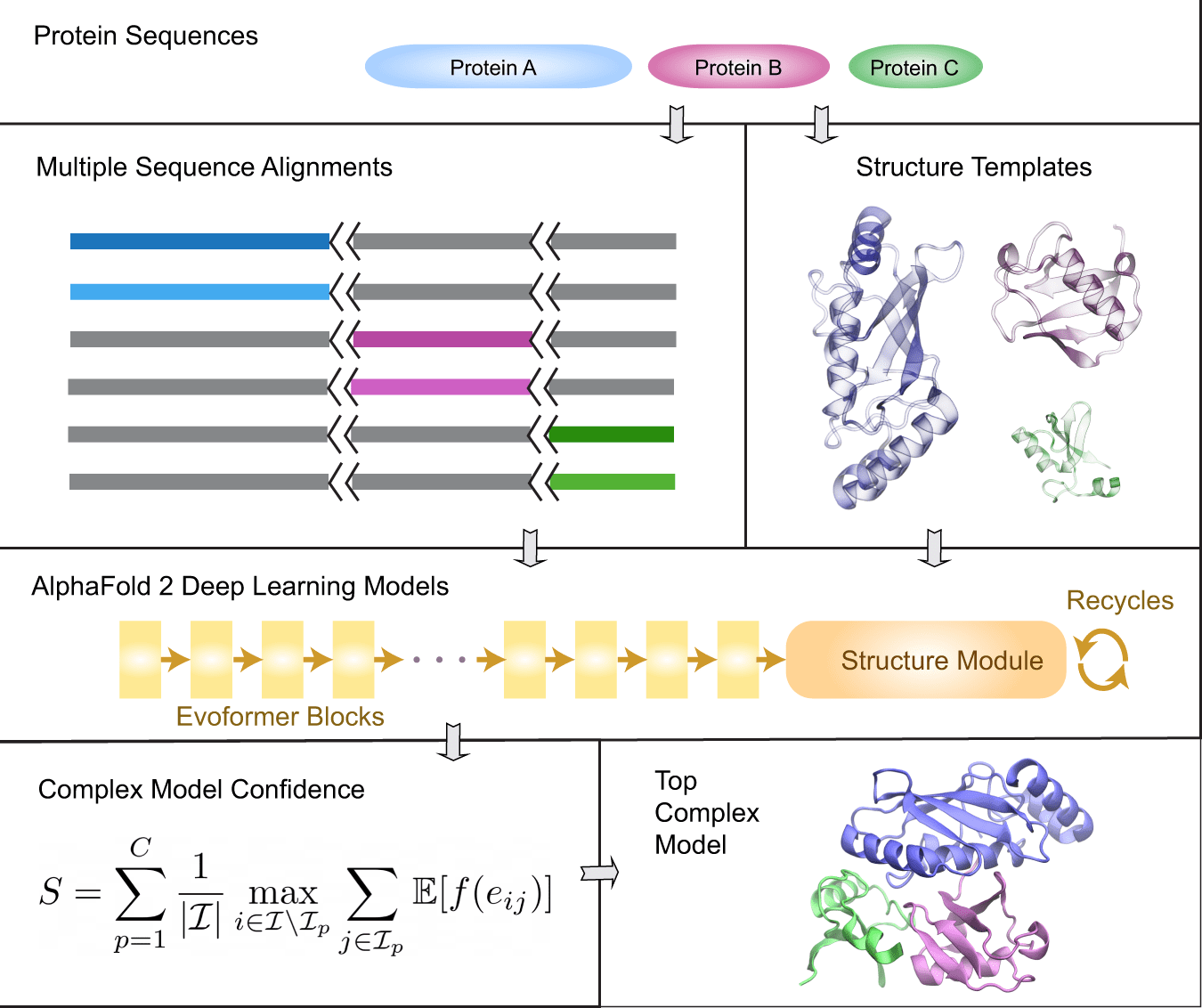
Image Source: AF2Complex predicts direct physical interactions in multimeric proteins with deep learning
AlphaFold2, a DeepMind-developed deep learning technique for predicting protein structure from a sequence, has significantly improved protein structure prediction. Because deep learning is a data-driven approach, the completeness of the structural space of single-domain proteins and the number of sequences in sequence databases are two significant aspects contributing to the success of AlphaFold2. These elements have been combined to allow sophisticated neural network models to be trained for accurate protein structure prediction. AlphaFold2 not only performed well on single-domain targets, but it also performed well on multidomain proteins and has been used for such proteins in various model organisms.
Several proteins that form complexes in prokaryotes are fused into long, single-chain, multidomain proteins in eukaryotes. The physical forces that drive protein folding are also associated with protein-protein associations. Understanding biological systems require accurate descriptions of protein-protein interactions.
AlphaFold2 recently computed incredibly accurate atomic structures for individual proteins. This study revealed that without retraining, the same neural network models created for single protein sequences in AlphaFold2 could be repurposed to predict the structures of multimeric protein complexes.
Is it possible to modify AlphaFold2 to anticipate the structure of a protein complex?
Following the release of AF2, the search for an explanation began almost immediately. One early work converted a two-chain structure prediction problem into a single-chain structure prediction problem by simply joining two protein sequences with a poly-glycine linker. A far better option is to change AF2’s “residue index” function, which avoids the requirement for a covalent linker, which is likely to cause artifacts.
Meanwhile, research has been conducted using docking methods and models of single proteins created with AF2. They are predicated on the assumption that AF2 provides high-quality monomeric models that could boost the likelihood of native-like docking positions. As some scientists pointed out, one problem with these findings is that the benchmark set utilized to train the AF2 deep learning models includes protein structures. The use of holo monomers in training compromises rigor because AF2 presumably gives an “observed” holo-structure for docking, despite the fact that the AF2 models were not trained on protein complex structures.
Is it possible to adapt AlphaFold2 to anticipate protein-protein interactions and identify higher-order protein complexes?
Several high-throughput experimental approaches have been developed to detect interacting protein partners, however, their results are typically incongruent and incomplete. Template-based techniques have been employed computationally, although they are limited to the discovery of homologs. Researchers have used a combination of classic protein-protein docking approaches, co-evolutionary signals, and even deep learning models on entire proteomes.
These are effective methods, but they need the use of paired multiple sequence alignments (MSAs) as inputs. The identification of orthologous sequences across species is required to generate paired MSAs, which is difficult in many situations due to the occurrence of paralogs in eukaryotes, protein cross-talk in disease pathways, and pathogen-host interactions.
In this study, the scientists show that AF2 can be tailored to predict both the presence of protein-protein interactions and the related quaternary structures using numerous test sets and without employing paired sequence alignments.
Deep learning model ‘AF2complex’
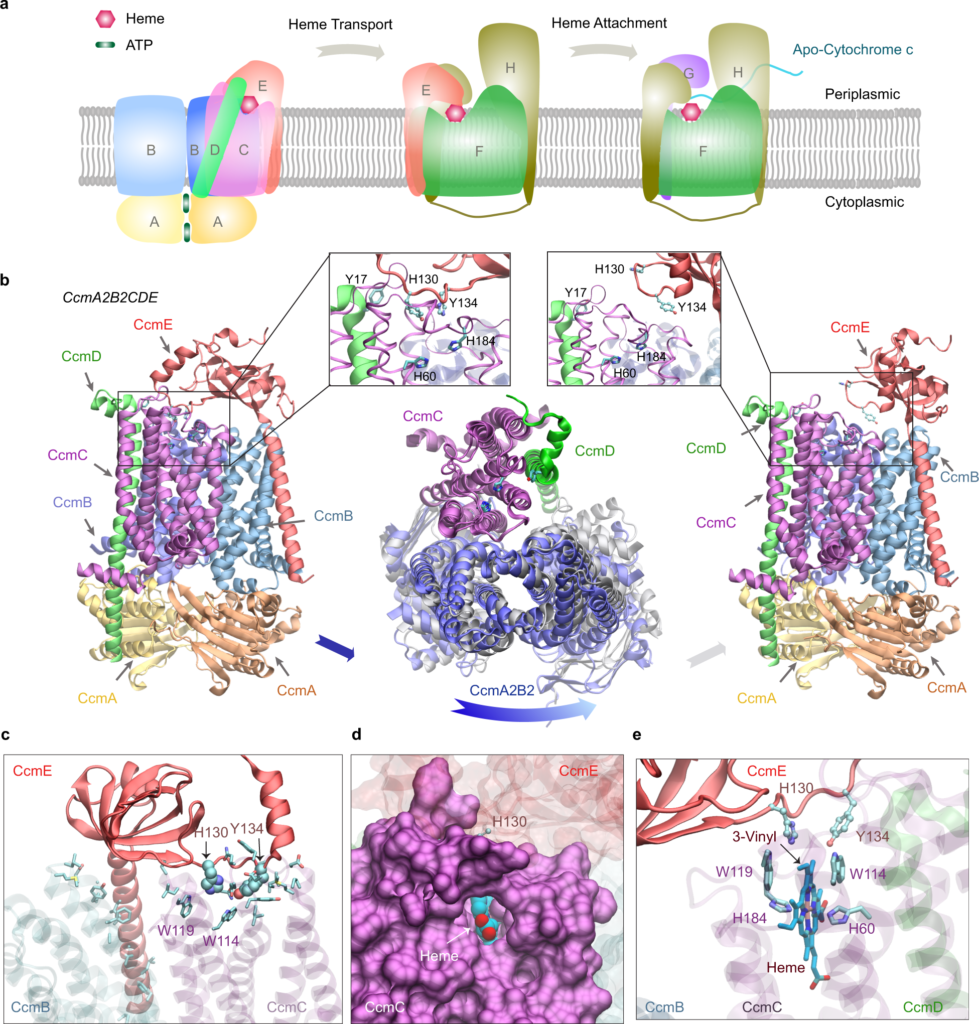
Image Source: AF2Complex predicts direct physical interactions in multimeric proteins with deep learning
For the provided query sequences of a target protein complex, the original AlphaFold2 data pipeline is first applied to collect input features for each query. Then, for complex structure prediction, AF2Complex assembles the individual monomer features.
AF2Complex, in contrast to other methods, does not require paired multiple sequence alignments. It outperforms several complex protein-protein docking techniques and AF-Multimer, AlphaFold’s multimeric protein creation.
The scientists also provide metrics for predicting direct protein-protein interactions between arbitrary protein pairs and test AF2Complex against various complicated benchmark sets as well as the E. coli proteome. Finally, in the case of cytochrome c biogenesis system I, the study presents high-confidence models of three sought-after assemblies formed by eight members of this system.
Final thoughts
In order to understand biological systems, it is essential to describe the interactions between proteins correctly. Recently, AlphaFold2 (AF2) computed atomic structures for individual proteins with remarkable accuracy. In this study, the authors demonstrate that multimeric protein complexes can be predicted using the same neural networks developed for single protein sequences. Moreover, it is more accurate than some complex protein-protein docking strategies and provides a significant improvement over AF-Multimer.
Story Source: Paper Reference | AF2Complex Code: GitHub | Zenodo
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











, to predict protein complex structure and protein-protein interactions.){kind=link}