
Scientists formulate a deep learning framework, DeepRank, for the data mining of 3D protein-protein interfaces. They demonstrated the efficiency and readiness of DeepRank on two unique difficulties in structural biology.
Image Source: DeepRank: a deep learning framework for data mining 3D protein-protein
3D structures of protein complexes give essential data to decipher biological processes at the molecular scale. The huge measure of experimentally and computationally resolved protein-protein interfaces (PPIs) offers the chance to train deep learning models to aid the predictions of their biological relevance.
The scientists present in the study, DeepRank, a generic, configurable deep learning system for data mining PPIs by the utilization of 3D convolutional neural networks (CNNs). DeepRank maps the features of PPIs onto 3D grids and helps train a user-specified CNN on the 3D grids.
DeepRank enables proficient training of 3D CNNs with datasets containing a large number of PPIs and upholds both classification and regression.
The scientists depict the performance of DeepRank on two distinct difficulties: The classification of biological versus crystallographic PPIs and the ranking of docking models.
For the two issues, DeepRank is competitive with, or outperforms, cutting-edge techniques, showing the versatility of the system for research in structural biology.
Gaining Knowledge of Interaction of Biomolecules
Profoundly regulated protein-protein interaction networks coordinate most cellular processes, going from DNA replications to viral invasion and immune defense.
Proteins interface with one another and other biomolecules in unambiguous ways. Acquiring knowledge on how those biomolecules interact in 3D space is key for grasping their functions and taking advantage of or engineering the molecules for a wide range of purposes, for example, drug design, immunotherapy, or designing novel proteins.
A Calling for Easily Modifiable Open-Source Frameworks
In the previous years, a range of experimental strategies (e.g., X-ray crystallography, nuclear magnetic resonance, cryogenic electron microscopy) have been used to determine and amass countless atomic-resolution 3D structures of protein complexes (>7000 non-redundant structures in the PDBe databank.
Various machine learning methods, and as of late, a few deep learning techniques, have been developed to learn complicated interaction patterns from the experimental 3D structures.
Unlike other machine learning methodologies, deep neural networks hold the guarantee of learning from a large set of data without arriving at a performance level rapidly, which is computationally tractable by reaping hardware accelerators (like GPUs, TPUs) and parallel file system technologies.
The scientists have trained 3D deep convolutional networks (CNNs) on 3D grids addressing protein-protein interfaces to assess the quality of docking models (DOVE). Apart from that, Geodesic CNNs have been applied to extract protein interaction fingerprints by applying 2D ConvNets on spread-out protein surface patches (MaSIF).
Graph Neural Networks (GNNs), which address protein interfaces as graphs, have also been applied to predict protein interfaces. At last, rotation-equivariant neural networks have, as of late, been utilized by scientists for point-based representation of the protein atomic structure to classify PPIs.
One exceptional delineation of the capability of deep neural networks in structural biology is the new leap forward in single-chain protein structure prediction by AlphaFold2 in the most recent CASP14 (Critical Assessment of protein Structure Prediction round 14).
Prediction of the 3D structure of protein complexes remains an open challenge: in CASP14, no single assembly was accurately predicted except when a known template was available. This calls for open-source systems that can be effectively altered and extended by the community for the data mining of protein complexes and can facilitate knowledge discovery on related scientific inquiries.
A Requirement for Generic and Extensible Deep Learning Frameworks
Data mining of the 3D protein complexes presents a few one-of-a-kind difficulties. To start with, protein interfaces are governed by physicochemical rules.
Various sorts of protein complexes (e.g., enzyme-substrate complex, antigen-antibody complex, etc.) may have different dominant attraction signatures. For instance, a few complexes might be driven by hydrophobicity and others by electrostatic forces.
Second, protein interactions can be classified at various levels:
- Atom-atom level,
- Residue-residue level, and
- Secondary structure level.
Third, protein interfaces are exceptionally diverse in shapes, sizes, and surface curvatures.
At last, efficient processing and featurization of an enormous number of atomic coordinates files of proteins is daunting with regards to computational expense and fike storage requirements.
Accordingly, there is an arising need for generic and extensible deep learning structures that researchers can undoubtedly re-use for their specific problems while eliminating tedious phases of information preprocessing.
Such generic systems have previously been developed in different scientific fields going from computational chemistry (DeepChem) to condensed matter physics (NetKet), and have fundamentally contributed to the rapid adoption of machine learning methodologies in these fields.
They have invigorated collaborative efforts, produced new bits of knowledge, and are persistently improved and maintained by their particular user communities.
DeepRank
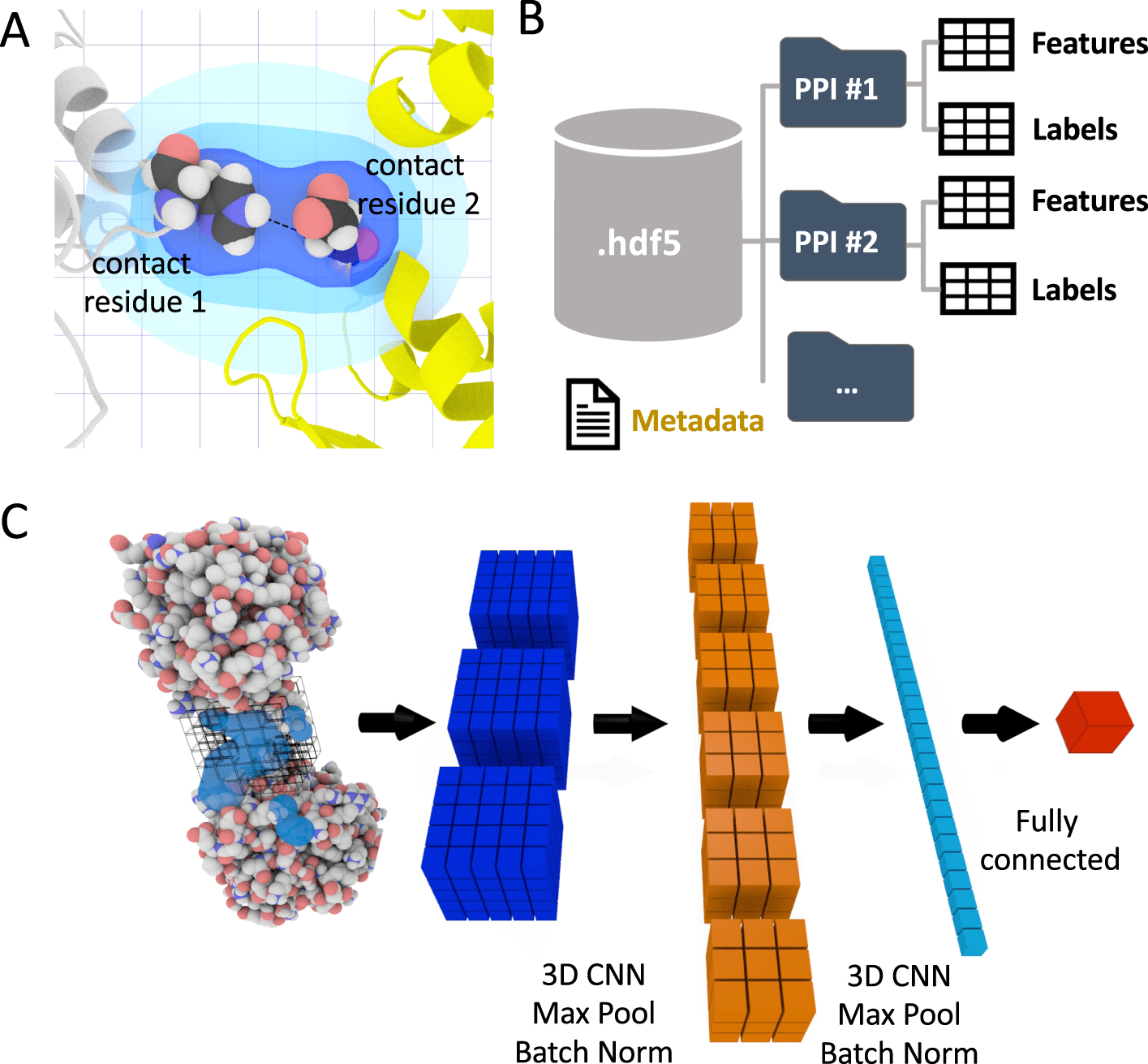
In the paper, the scientists present DeepRank, a generic deep learning platform for data mining protein-protein interfaces (PPIs) in light of 3D CNNs. DeepRank maps atomic and residue-level features determined from 3D atomic coordinates of biomolecular complexes in Protein Data Bank format onto 3D grids.
DeepRank applies 3D CNN on these grids to learn problem-explicit interaction patterns for user-characterized tasks. The architecture of DeepRank is profoundly modularized and optimized for high computational efficiency on extremely large datasets up to millions of PDB files.
It permits users to characterize their own 3D CNN models, features, target values (e.g., class labels), and data augmentation strategy. The platform can be utilized both for classification, e.g., predicting an input PPI as biological or a crystal artifact, and regression, e.g., prediction of binding affinities.
DeepRank is built as a Python 3 package that permits end-to-end training on datasets of 3D protein-protein complexes.
The overall structure of the DeepRank package comprises two principal parts, one focussing on information preprocessing and featurization and the other on the training, assessment, and testing of the neural network.
The featurization takes advantage of MPI parallelization along with GPU offloading to guarantee proficient computation over exceptionally large data sets.
The scientists initially defined the structure of their DeepRank framework. To exhibit its applicability and potential for structural biology, they applied it to two different research challenges.
The researchers first presented the performance of DeepRank for the classification of biological versus crystallographic PPIs. With a precision of 86%, DeepRank outperforms cutting-edge strategies, such as PRODIGY-crystal and PISA, which reach an accuracy of 74 and 79%.
The researchers then, at that point, present the performance of DeepRank for the scoring of models of protein-protein complexes generated by computational docking. They demonstrated that DeepRank is competitive and some of the time outperforms three best-in-class scoring functions: HADDOCK, iScore, and DOVE.
The Endpoint
In conclusion, scientists have defined an open-source, generic, and extensible deep learning system for data mining exceptionally large datasets of protein interfaces. They demonstrated the efficiency and readiness DeepRank on two unique difficulties in structural biology.
The scientists predicted that DeepRank would accelerate scientific research connected with protein interfaces by facilitating the tedious steps of information preprocessing and diminishing daunting computational costs that might be related to large-scale data analysis.
The platform’s modularized and extendable system bears incredible potential for stimulating collaborative advancements by the computational structural biology community on other protein structure-related topics and will contribute to the adoption and development of deep learning strategies in structural biology research.
Article Source: Renaud, N., Geng, C., Georgievska, S. et al. DeepRank: a deep learning framework for data mining 3D protein-protein interfaces. Nat Commun 12, 7068 (2021). https://doi.org/10.1038/s41467-021-27396-0
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}