
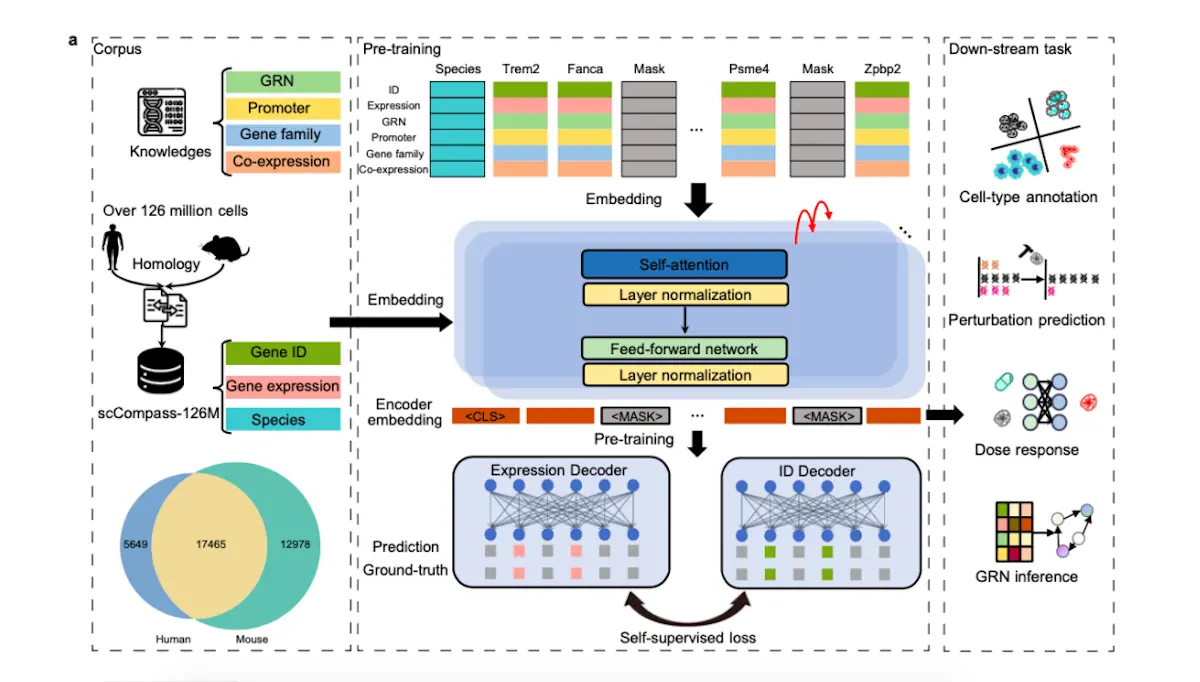
A team of researchers from the University of Chinese Academy of Sciences, China, and collaborators developed GeneCompass, one of the first foundation models of its kind that encompasses a vast expanse of knowledge across a diverse array of species, owing to the fact that it has been trained on over 120 million single-cell transcriptomes derived from the genomes of mice and humans. It is a self-supervised model. During the process of pre-training the model, it retrieves information from four types of biological datasets in the form of ‘prior knowledge’ and integrates it. It has excelled and outperformed several state-of-the-art models when studying a single species. It can also open new avenues for carrying out studies across different combinations of species other than humans and mice. This model can potentially contribute to discovering key regulators that determine cell fate and to identifying promising target candidates in the drug discovery and development field.
Why should we undertake studies at a cellular level?
It is essential to decode universal regulatory mechanisms that dictate the expression of genes across a diverse set of organisms for accelerating clinical research and expanding our existing knowledge of basic and crucial life processes. Traditional research methodologies and existing deep-learning models have only considered using individual models of organisms separately. This has resulted in a dearth of integrated knowledge of features observed across different cell types over a variety of species. The development of this model was made possible by combining the outcomes of recent advancements in the fields of deep-learning (DL) methods and single-cell sequencing methods.
Trillions of cells reside within vertebrate organisms, sorted into complex systems with many classifications. They are grouped to form functional tissues, which further clump together to form specialized organs that have specific physiological roles to facilitate the survival of the organism they lie within. Developing a sound understanding of the universal regulatory mechanisms common to all these cells, tissues, and organs is necessary for recognizing patterns observed during the development of an individual. Many regulatory mechanisms have been conserved throughout evolution, and it is in the interest of researchers to study these alongside the diversity and variation caused by evolutionary processes. This, in turn, contributes to curating clinical therapeutic methods accordingly.
The importance of single-cell data
The accumulation of a plethora of single-cell data has been made possible due to developments in omics sequencing methods; this data can help researchers understand specific functions of specialized cells within their respective organs, down to pinpointing their activities within just a single cell. The regulation of gene expression can be controlled by factors such as post-transcriptional modifications and by assessing the accessibility of chromatin.
Deep-learning methods aid the process
Using wet-lab experiments to decipher regulatory mechanisms across such a large scale of cells is very time-consuming and requires extensive labor, which in turn increases the operation costs as well. Deep-learning methods can recognize complicated patterns across large datasets. It is the light at the end of the tunnel that has been searching for efficient and quick methods for analyzing the regulatory mechanisms common to millions and trillions of cells.
Existing foundation deep-learning models
In the visual domain, DALL-E is a popular foundation model, and for natural language applications, we have BERT (used for predicting peptides and codons), GPT (for example, ChatGPT), and LLaMA. These are considered state-of-the-art (SOTA) models in their respective domains of implementation.
Pre-training takes place when models learn in a self-supervised manner. Once that is done, the model adapts specific steps to carry out downstream tasks that have been assigned to it; this process is known as fine-tuning. All models require a representative layer for fine-tuning; for GeneCompass, transcriptome data fulfills this role. This layer represents regulatory activities for genes that are located in different biological systems.
Some examples of pre-existing deep-learning models that use single-cell data are Geneformer and scGPT. They can perform downstream tasks like predicting target drugs, annotating cell types, and clustering cells. Their only major limitation is that they depend on data retrieved from just a single species.
GeneCompass enters the picture
It has been pre-trained on data retrieved from scCompass-126M. ‘Prior knowledge’ is a term that includes the following:
- Annotations of different families of genes
- Interactions between genes that have been verified experimentally
- Data on core regions of regulation across the genome
- Regulatory relations between transcription factors and their targets
- Networks of co-expressing genes
- Promoter sequences
This model surpasses SOTA models in terms of performance. To enable a better contextual understanding, self-attention mechanisms are used. Expressive genes that provided extensive information and showed high variability were retained in the training datasets to account for biological signatures specific to different cell types and to acknowledge cell heterogeneity. The researchers involved in this study investigated the effect of the assembly of the aforementioned ‘prior knowledge’ on tasks involving annotation and the incorporation of different types of cells by carrying out several experiments. The results proved that prior knowledge positively impacted the performance of the model. They also showed that the model’s abilities to decode gene functions at extremely high resolutions could be further increased by adding gene expression values along with information on gene rankings, as opposed to using them separately, which has been the case with previous models. It can also screen genes that promote the directed differentiation of cells.
Conclusion
This study took only two species, humans and mice, into consideration. Another issue was the introduction of data related to gene expression patterns for specific species; it overshadowed the advantages of using single-cell data. Other important aspects, like the effects of enhancers and protein sequences, have not been incorporated into the ‘prior knowledge’ dataset.
There is further scope to include rich data obtained from proteomic, epigenomic, and metabolic studies, as they provide valuable insights into gene regulation. There is also potential for the model to contribute to the optimization of systems that reprogram cell fate, direct the cultivation of organoids, predict the toxicity of drugs, and screen drugs for their corresponding tumors. There are hopes and expectations that this model can make the analysis of results obtained from wet lab experiments much more efficient and quick by combining both dry lab (in-silico) and wet lab (in-vitro) data.
Article source: Reference Paper
Important Note: bioRiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.











{kind=link}