
Scientists from A*STAR’s Genome Institute of Singapore (GIS) discovered that carbapenemase-producing Enterobacteriaceae (CPE) could hide out among gut bacteria in asymptomatic humans. Long-term research to understand microbiome ecology within the population suggests visible shifts in microbial pathways assisting in the colonization of Enterobacteriaceae favoring CPE (Carbapenemase-producing Enterobacteriaceae ) infections. The study provides imperative insight into future interventions to curb the AMR burden associated with these colonizing bacteria.
Antimicrobial resistance (AMR) and associated genes have been a major threat to public health dissemination to understand community transmission and address potential interventions. The research group led by computational biologist Niranjan Nagarajan at Genome Institute of Singapore, alongside clinicians, reviewed CPE infections associated with asymptomatic gut colonization. Scientists primarily focused on the cohort of patients who have undergone decolonization and evolutionary changes associated with the transmission of resistance genes in the gut microbiome.
The study highlights its work by pressing the favored history of carbapenemase-producing Enterobacteriaceae, which includes species like Escherichia coli and Klebsiella pneumonia, being resistant to the last line of antibiotics. The longitudinal study utilizes techniques such as deep metagenomic sequencing, whole genome sequencing, and sub-strain gene variation to highlight targeted intervention in-vitro studies on the strain and genetic levels to reduce AMR burden and colonization competition against commensals.
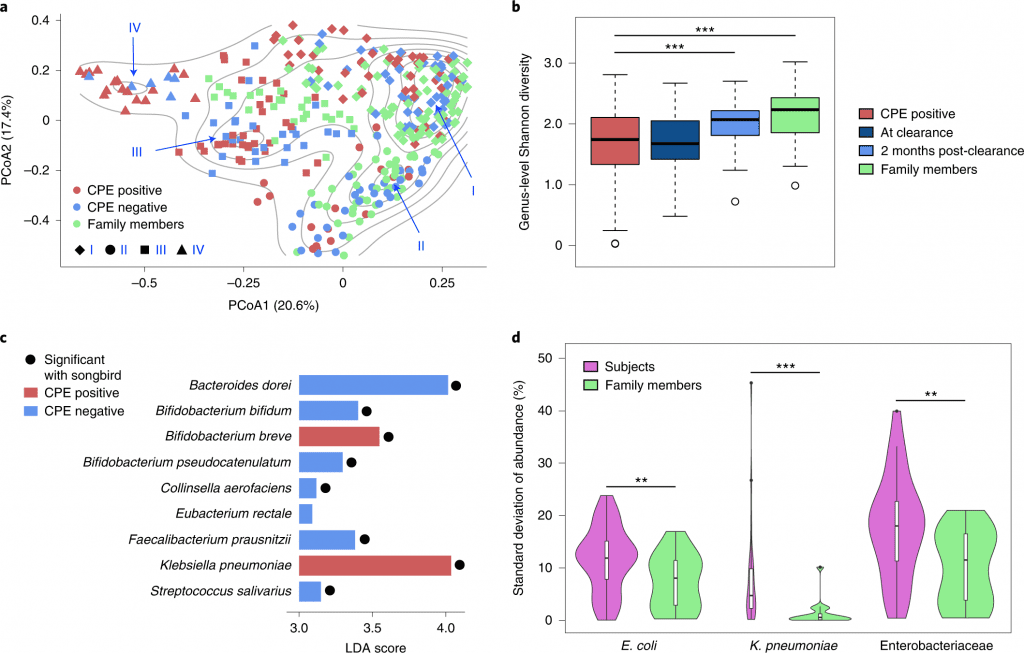
Image Source: https://doi.org/10.1038/s41564-022-01221-w
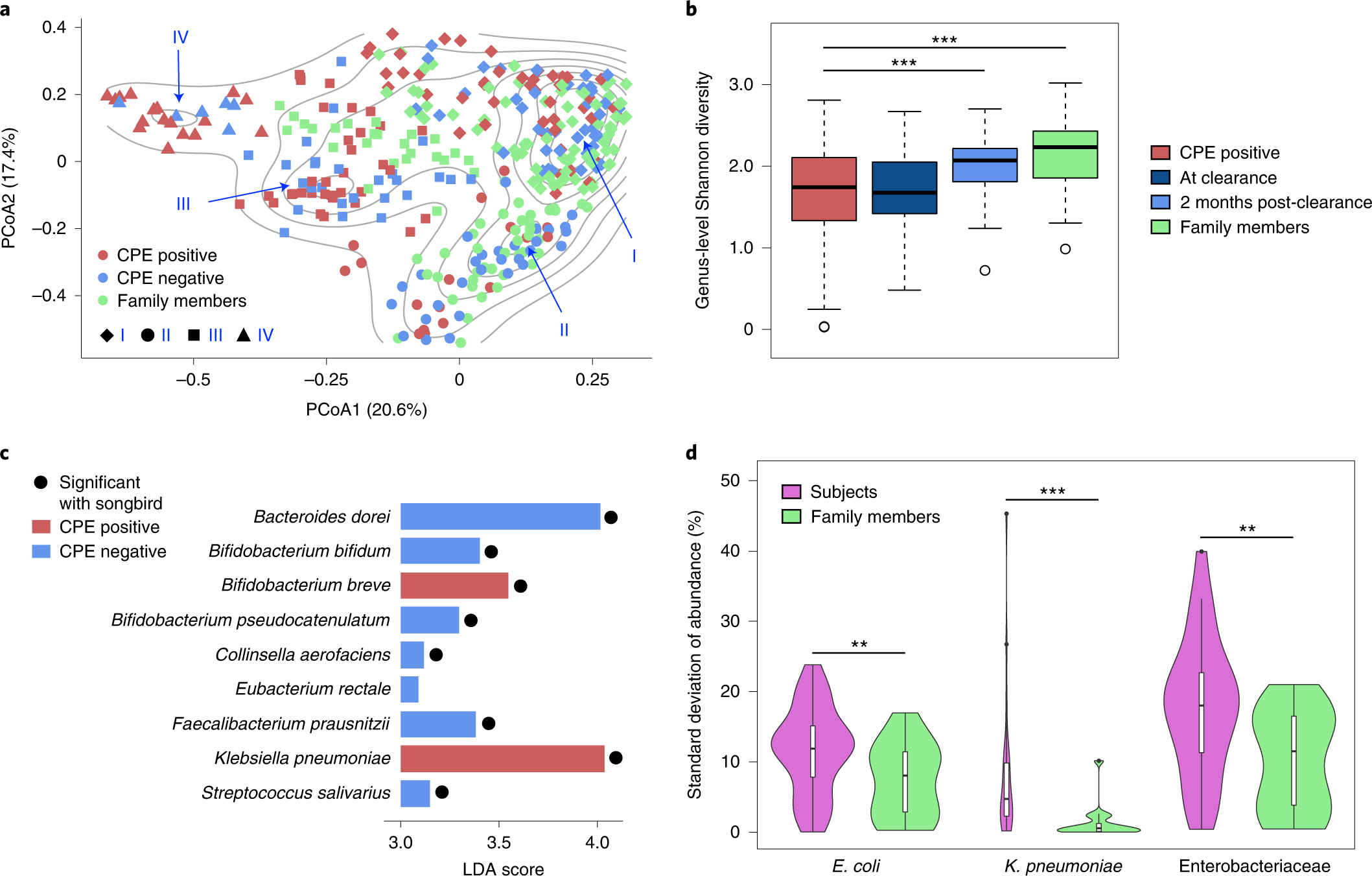
The cohort was based on subjects who had undergone decolonization and their respective family members to have a home environment matched group as controls. A total of 361 fecal samples were obtained over two years. Comparative analysis based on shotgun metagenomic analysis and whole genome sequencing data revealed shifting of microbial communities across the gut ecology of patients positive for CPE genes (blaNDM-1, blaKPC, blaOXA-48, blaIMI-1 and blaIMP) also resistant to antibiotic drugs ertapenem or meropenem. Followed by genus-level Shannon diversity, which displayed a gradual increase in microbial diversity within CPE-positive patients post decolonization and healthy controls.
As a result, researchers extended work to identify species associated with CPE infections post decolonization, and Klebsiella sp. was largely enriched in individuals. Only one commensal species, Bifidobacterium breve, was enriched and could be of importance in alleviating inflammatory pathways associated with infection. The differential pathway analysis was conducted to conclude that metabolic processes such as sulfur and nitrogen pathways as well as oxygen homeostasis were favorable for Enterobacteriaceae species due to the abundance of a micro-aerophilic environment.

Image Source: https://doi.org/10.1038/s41564-022-01221-w
To identify strain-specific dynamics of the CPE species, single nucleotide variant analysis was used to determine the frequency of strains that predominantly colonize the gut, E.coli showed similar prevalence patterns in both subjects (86%) and healthy controls (90%), while K. pneumonia was more frequent in subjects (70%) relative to family members (30%). These results highlight the differences based on strain-specific dynamics, which explains the association of pro-inflammatory responses within the gut. The sub-strain variation was analyzed to determine single strain virulent genes to understand the evolutionary mutations. Interestingly, the CPE positive cohort was identified with multiple clusters (distinct SNV- dnaK, the 30S, and 50S ribosomal subunits) while several genes were common to both E.coli and Klebsiella species.
Considering the transposability of antimicrobial genes within the CPE infection, the team also studied the plasmid sequence analysis and identified two synergistic common plasmids within CPE bacterial isolates. Between the two, pKPC2 (carbapenem-resistant Klebsiella pneumonia species) was shown to have conjugation post 300 generations of plasmid clones bearing blaKPC-2, the carbapenemase. The study deduced and allowed introspection of long-term dynamics, enabling gut microbiome comparisons for CPE decolonization before and after. Comparing the cohorts with the family control group distinctly recapitulated the shifting of the associated microbial consortium with decolonization.
Final Thoughts
The study revealed how CPE microbial strains do not alone account for colonization in the gut microbiome. The exploitation of antimicrobial drugs in various classes throughout the years has posed a threat to public health. The swift shifting of microbial pathways and metabolism of the gut bacteria largely favors their growth during gut inflammation and other pro-inflammatory responses. Future perspectives for interventional therapeutics for probiotic formulations could prove beneficial due to the prevalence of commensal species.
Article Source: Reference Paper | Data availability: ENA | Code availability: GitHub
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Mahi Sharma is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She is a postgraduate with a Master's in Immunology degree from Amity University, Noida. She has interned at CSIR-Institute of Microbial Technology, working on human and environmental microbiomes. She actively promotes research on microcosmos and science communication through her personal blog.





 could hide along gut microbiome in asymptomatic humans.){kind=link}