
A large percentage of the global population is shown to have been affected by different types of cancer and increased mortality. Hepatocellular carcinoma, or primary liver cancer is one the life-threatening disease and the fastest-growing cancer type in the United States. A recent study conducted by Hidewaki Nakagawa et al. leveraged the proteomic profiling of primary liver cancer to decipher potential subtypes and therapeutic strategies.
Introduction
In recent years, there has been a constant increase in cancer incidence and mortality rate in the global population. With the highest mortality of the population surrounding virus-associated liver cancers. Several research studies in recent years have elucidated the heterogeneity of liver cancer in terms of disease etiology, histology, and therapeutic responses. Considering the heterogeneity of the medical condition, it often harbors difficulty in assisting medical responses. Hence, the large-scale molecular characterization and proteomic profiling of virus-associated liver cancer can provide insights into personalized cancer treatments.
Image Source: https://doi.org/10.1038/s41467-022-34249-x
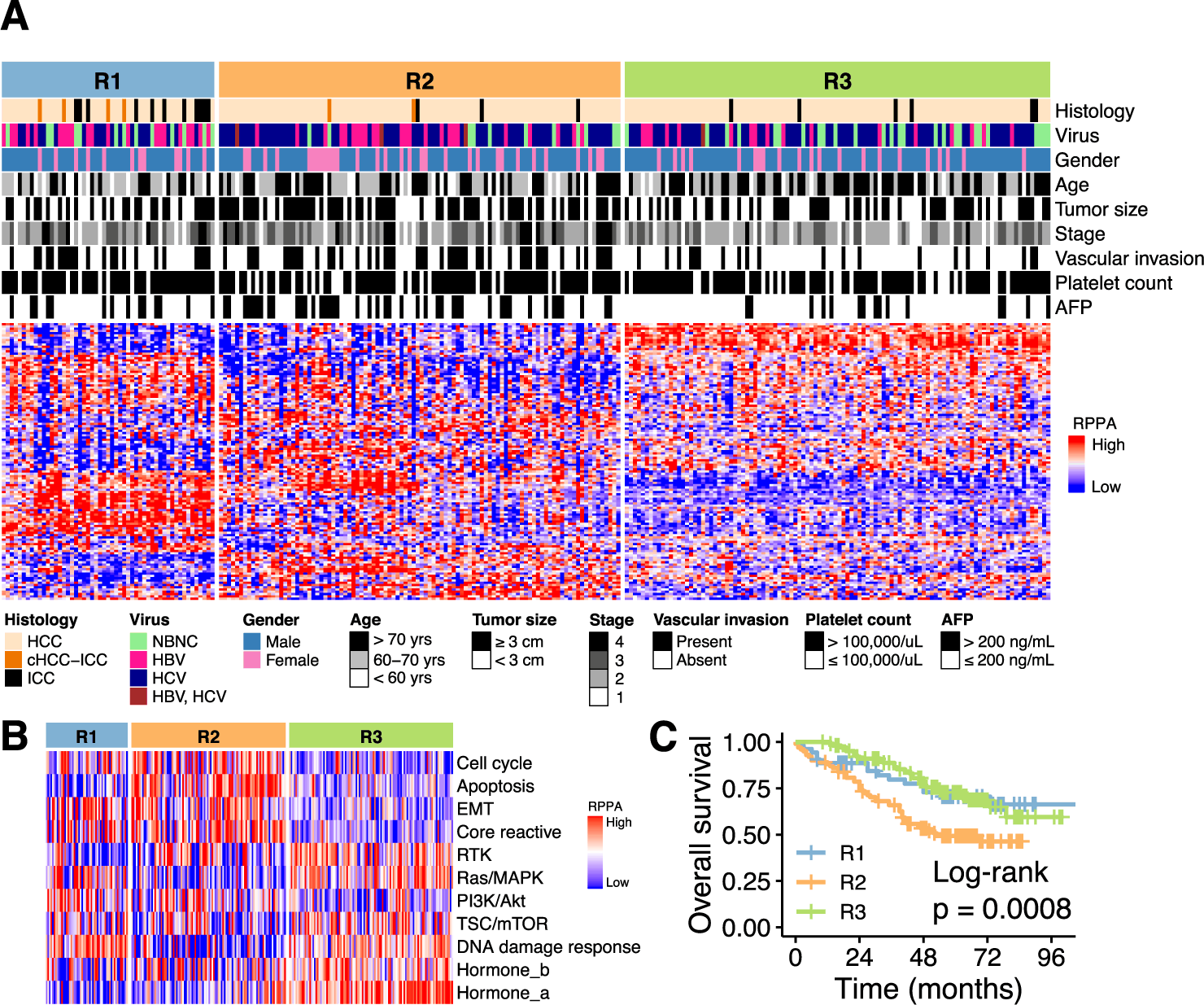
A team of researchers associated with the RIKEN Center for Integrative Medical Sciences, Yokohama, Japan, and The University of Texas MD Anderson Cancer Center, Houston, TX, USA, have addressed the heterogeneity of the associated liver cancer by leveraging the proteomic-genomic profiling using reverse-phase protein array (RPPA). The histology of liver cancer is categorized as hepatocellular carcinoma (HCC), intrahepatic cholangiocarcinoma (ICC), and cHCC-ICC (combined hepatocellular carcinoma-intrahepatic cholangiocarcinoma) arranged in the decreasing recurrence.
The potential of proteomic profiling in clinical tumor tissues can help decipher the molecular characteristics and correlate disease for personalized therapy. Several studies conducted by researchers across the globe have analyzed the liver tissues resulting from HBV (hepatitis B virus) and HCV (hepatitis C virus ) using approaches such as mass-spectrometry-based global proteomics by Jiang et al. that resulted in 110 early-stage HCC identification aided with an average of 5953 proteins per tumor. Consequently, the research conducted by Gao et al. and Dong et al. resulted in assessed HBV-associated HCC from 159 patients, identifying on average, 8934 proteins per sample, whereas proteomic profiling on 214 ICC liver tissue found 7385 proteins per sample.
Although the earlier research studies provide an unbiased insight into liver cancer proteome, the limiting factors affecting the analysis, such as both sample size and quantitative analysis for the samples fairly limited the implications. Considering the potential of RPPA, providing high throughput and cost-effective technology for measuring the protein expression levels on a large scale of samples. The current study employs the proteomic profiling of approximately 300 proteins on the 259 primary liver cancer positive for HCV and HBV viruses using the RPPA to decipher potential subtypes of cancer using unsupervised classification and insights into targets for therapeutic potential.
Hidewaki Nakagawa et al. and team based the research study on a diverse population using samples of Japanese ethnicity. The team examined the protein abundance correlation with the mRNA abundance, protein abundance was generally positively correlated with mRNA abundance. Within the 222 mRNA-protein pairs, 30 pairs (14%) had negative correlation coefficients with potential reasoning, such as some of these proteins include low-expression levels, low concentration range, heterogeneity of the tissue, and regulation of the post-transcriptional process. Researchers extended the understanding of correlations in cancer cohorts of TCGA using mRNA-protein pairs for similarity in levels resulting in the highest parallel of the TCGA liver cancer cohort (LIHC) and Hidewaki Nakagawa and team cohort.
Considering the impact of both mRNA and protein abundance in reference to somatic copy number alterations (SCNAs), researchers examined a total of 209 mRNA protein pairs for their association with the SCNAs. The results implicated various distant interactions between mRNA and SCNA on contrasting chromosomes. Approximately 30 of 209 proteins (14%) had significant local interaction with the SCNA, including 1p, 1q, 4q, 5q, 9, 10, and 22 that were discovered in the RNA interactions.
In order to obtain insights into the heterogeneity of virus-associated clinical liver cancer, researchers performed a hierarchical clustering of 259 tumors based on protein abundance. The clustering identified three subclasses for the tumor; hence, three subclasses R1, R2, and R3, classified 259 tumors as 53, 100, and 106 respectively. The proteomic subclasses were observed to have various associations with clinical features such as the R1 subclass was younger and posed a good prognosis of cancer, the R2 subclass displayed an advanced form of the proliferative tumor with TP53 mutations with the worst prognosis and increased expression of VEGF-2, and R3 subclass was observed to be enriched in mutations such as CTNNB1 and elevated activity of mTOR signaling pathway.
In order to identify the potential somatic driver alterations of the liver cancer samples, the team analyzed samples for whole genome sequencing to discover the genomic and transcriptomic features of the proteomic subclass. The analysis resulted in TP53 and CTNNB1 mutations being enriched in the R2 and R3 proteomic subclasses respectively. The therapeutic implication of drugs within the proteomic subclass resulted in the R2 subclass selectively responding to bevacizumab or multi-kinase inhibitor (MKIs) with a selective inhibitory effect on VEGFR2, R1 subclass is regarded as an immunologically “hot” tumor and may constitute a patient cohort that would selectively aid from atezolizumab or other immune checkpoint inhibitors while R3 subclass contained lower levels of immune infiltrates, VEGF signaling suggesting a new approach for this subclass.
The research team examined 293 RPPA-measured proteins for the association of potential prognostic markers since protein-based prognostic biomarkers have clinical applications. In R1 and R2 subclasses (SRC, tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein ζ (YWHAZ), lysine acetyltransferase 2A (KAT2A), fibronectin 1 (FN1), and TYRO3 protein tyrosine kinase (TYRO3)) and three (transferrin receptor (TFRC), ribosomal protein S6 (RPS6), and PCNA) were largely enriched whereas ULK1-pSer757, hexokinase 2 (HK2), membrane-spanning 4-domains A1 (MS4A1), glycogen synthase 1 (GYS-pSer641), CDKN2A, PGR, and DUSP4 were abundant in the R3 subclass.
Future Perspective
The current study performed large-scale proteomic profiling of primary liver cancer using RPPA. Analyzing approximately 259 tumor tissues to understand the heterogeneity of cancerous tissues on liver cancer proteome. Since the cohort mainly constituted a population that is relatively HCV-positive patients of Japanese ethnicity it clearly outpasses earlier research and builds understanding about the proteomic subclasses R1, R2, and R3 and genomic insights into them. The team has successfully presented a large dataset of liver cancer proteome and genome, which allows an exploration of molecular heterogeneity among primary liver cancers and provides direction for personalized therapeutics.
Article Source: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Mahi Sharma is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She is a postgraduate with a Master's in Immunology degree from Amity University, Noida. She has interned at CSIR-Institute of Microbial Technology, working on human and environmental microbiomes. She actively promotes research on microcosmos and science communication through her personal blog.





{kind=link}