
A team of researchers from McGill University and Hunan University developed MiRGraph, a multi-view feature-learning method based on transformers that have the ability to improve miRNA-target interaction (MTI) predictions by modeling the features of sequences in heterogeneous networks. Gene expression is regulated by microRNA (miRNA), which are small single-stranded RNA molecules present in the cell. Messenger RNA (mRNA), which codes for the protein to be produced post-translation, is targeted by miRNA. Based on which miRNA targets the mRNA, mRNA molecules can either act as oncogenes (genes that promote the proliferation of cancerous tumors) or as tumor suppressors. Studying the interactions between miRNA and their targets is referred to as miRNA-target interactions (MTIs); it helps us understand the complexities involved in gene regulation as well as in determining diagnostic markers and potential targets that can be used in medical therapy. Currently, there haven’t been enough studies that involve feature learning on sequence information and heterogeneous graph networks for MTI prediction.
An introduction to miRNA
MicroRNAs are small, single-stranded molecules of RNA that do not code for any genes, i.e., they are non-coding molecules located within living organisms. On average, they contain between 22 and 23 nucleotides and are intricately involved in biological activities such as transcription, proliferation, and differentiation of cells within humans, as well as in regulating the immune response.
MiRNA induces the degradation of mRNA by binding to untranslated regions (UTRs) present at the 3’-end of the mRNAs that they are targeting; thus, miRNA acts as an important regulator for post-transcriptional activities. Any problems that come up during gene regulation by miRNA can have severe consequences, such as the proliferation of different types of cancer. Therefore, in order to treat diseases like cancer more effectively, it is imperative that we have a solid understanding of the complexity involved in gene regulation.
Traditional methods of capturing MTIs
Microarray experiments and qualitative real-time PCR (qPCR) have proven to be state-of-the-art methods for encapsulating MTIs and the regulatory functions associated with them. A drawback of these methods is their high cost and long duration of processing data, making them inconvenient to use on an industrial scale. Another problem arises from the fact that new miRNAs are constantly being discovered at an increasingly rapid pace, and frequently updating these technologies to keep up with this rate is simply unfeasible. This calls for developing alternative methods to select MTIs for validating experimental procedures; therefore, researchers have looked towards computational approaches to solve this issue.
Computational approaches for predicting MTIs
Two types of computational features exist for predicting MTIs: methods based on feature extraction and deep learning.
- Feature-based extraction methods
These methods contain features that are exclusively hand-crafted and meant for specialized purposes. However, this can act as a double-edged sword; while feature-based functions give candidate targets as their output, these methods are very dependent on engineering strategies for which the features have been hand-crafted, leading to many inconsistencies in predictions.
- Deep-learning methods
Algorithms like DeepTarget, TargetNet, and miRAW have used miRNA candidate-target sites (CTSs) to decrease the space that needs to be searched by the models. These models are heavily reliant on sequence information related to RNA and, therefore, do not make efficient use of information on miRNA-regulatory networks. As an alternative to extracting information related to the relationship between two different species of RNA, graph neural networks have been utilized using miRNA-mRNA interaction databases like StarBase and MirRTarBase that have accumulated relevant information related to these interactions throughout the years. However, even these methods have warranted improvements in the form of integrating node information into heterogeneous graph networks simultaneously.
Introducing MiRGraph
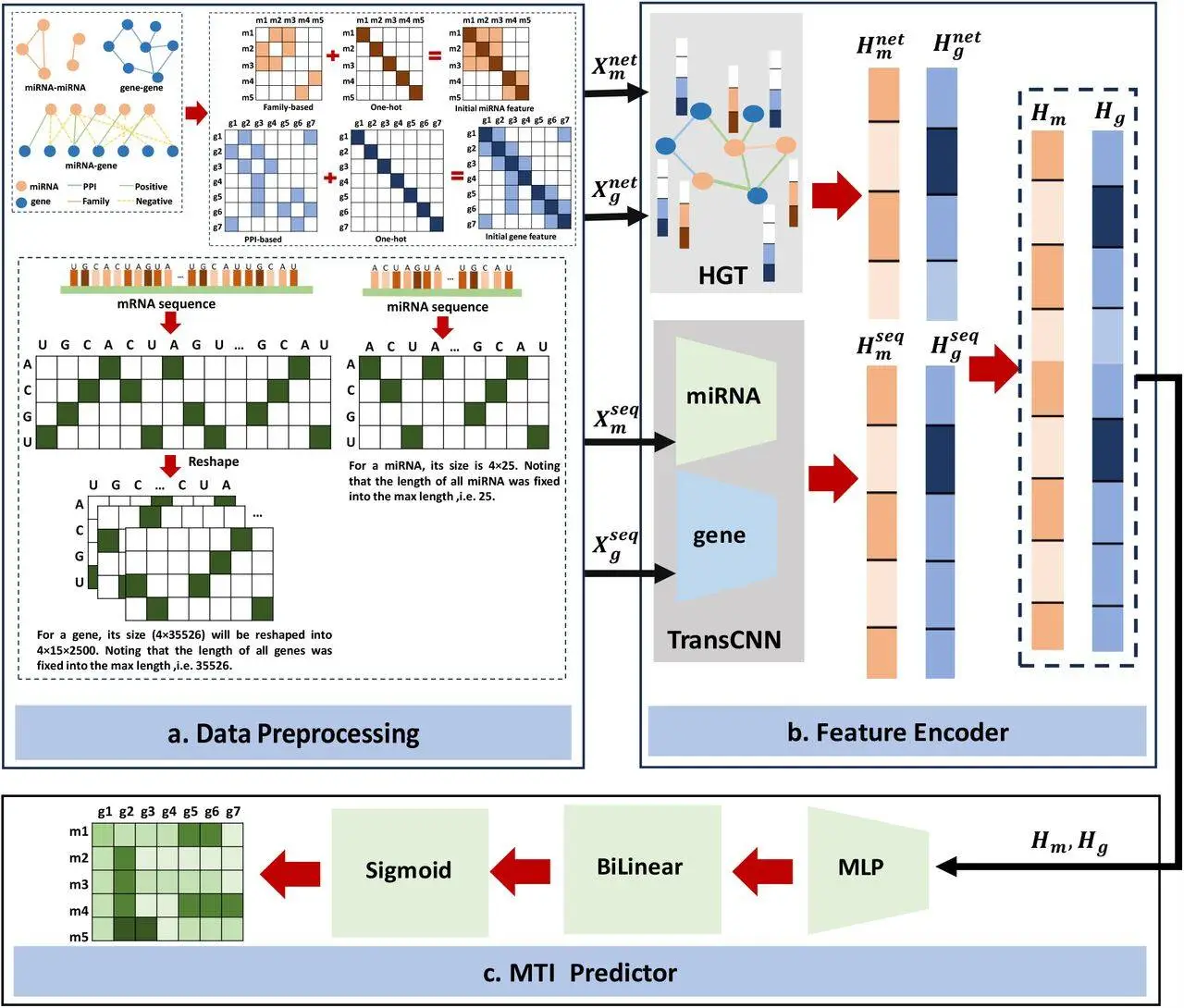
This model takes both sequence information and networks into account when making MTI predictions. Mature miRNA sequences are utilized to prevent the possibility of any sequence information that could be partially missing when considering k-mer encodings or candidate-target sites (CTSs). For personalized features to be extracted from miRNA molecules and genes, respectively, a module known as TransCNN has been designed by the researchers. It is a convolutional neural network (CNN) based on transformers and is used separately for miRNA and mRNA. It addresses the differences in sequence lengths for both molecules.
The sequential features are initialized by the model using information on raw sequences of mature miRNA and untranslated regions at the 3′ end of target mRNAs. The structural and relational information related to miRNA-miRNA, miRNA-target, and gene-gene interactions is all present within a heterogenous graph; to learn all the network features, the researchers used a heterogenous graph-transformer model.
To calculate MTI prediction scores, a bilinear function was used; on the other hand, to map the features of miRNA that have already been learned by the model into the same space, a multilayer perceptron (MLP) was adopted by the researchers. The MLP model was used after combining the features of miRNA and genes in the same embedding space. Topological features were captured using a heterogenous graph transformer (HGT) to understand relational and structural information within the heterogenous network in a more comprehensive manner. Additionally, to train the model, a focal loss function was used to prevent any imbalances between negative and positive samples. The researchers used qualitative methods of analysis to evaluate the capacity with which MiRGraph can differentiate high-functional MTIs and identify new MTIs.
MiRGraph has proved to be better than existing deep learning algorithms for making MTI predictions based on multiple standard metrics evaluated by the researchers. The field of biological research and development can benefit from the identification of innovative and highly functional MTIs in a number of ways.
Conclusion
The design of MiRGraph is similar to Enformer, an algorithm made for the purpose of predicting chromatin profiles from given sequences of DNA; however, it has been incapable of interpreting network-graph-related information. The researchers also mentioned how MiRGraph was used to find hsa-miR-181a-5p, a potential oncomir. In subsequent iterations of this model, they intend to integrate miRNA’s interactions with other cell constituents, including those with genes and circular RNA (circRNA).
Article Source: Reference Paper | MiRGraph is implemented in Python and available on GitHub
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.





{kind=link}