
The search for molecules that bind effectively to specific proteins is crucial from a drug discovery point of view. The existing methods for creating these chemicals are time-consuming and costly. In this study, TargetVAE, a one-of-a-kind AI model developed by researchers from Indiana State University, is utilized to generate potential drug-like molecules with high binding affinities to any protein structure. In contrast to earlier techniques, TargetVAE incorporates different protein representations, such as amino acid sequences and 3D structures, using a unifying framework called Protein Multimodal Network (PMN). This allows the model to generate a large number of ligand candidates that can bind to different portions of a protein without knowing the exact binding sites ahead of time. The model generation process is faster, and no unique property networks are required for each target protein.
Furthermore, pretrained AI models are used in the study to diversify the synthesized compounds while assuring relevance to the target protein. Overall, TargetVAE and PMN exhibit excellent results in terms of creating potential therapeutic candidates, demonstrating a substantial improvement in drug development through the integration of artificial intelligence. The software generated as part of this work is freely available to the public, which contributes to the scientific community. Furthermore, the study uses pre-trained AI models to diversify the chemicals synthesized while ensuring relevance to the target protein. Overall, TargetVAE and PMN provide great prospective therapeutic candidates, suggesting a significant advance in drug discovery via the use of artificial intelligence.
Variational Auto-Encoders (VAE)
A variational auto-encoder is a model used in complicated data processing, particularly in medicine development. It’s made up of two parts: a generative model that generates data and an inference model (probabilistic encoder) that interprets the data’s underlying structure. Using a decoder, the generative model generates data from a set of latent variables and a prior distribution. The goal is to maximize the log-likelihood of the data, but this is challenging due to the difficulty of considering all possible variations in the data. Variational auto-encoder addresses this by employing an encoder to approximate the distribution of the latent variable and maximize the Evidence Lower Bound (ELBO) during training.
Protein Multimodal Network (PMN)
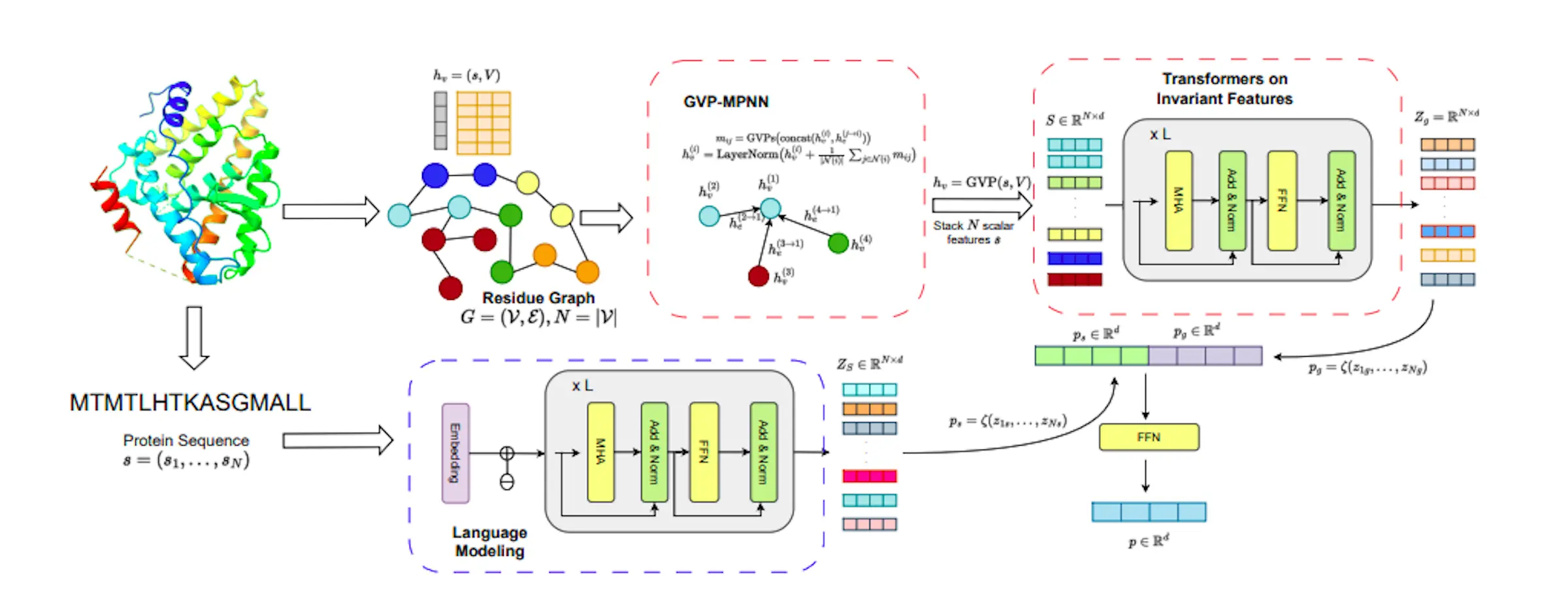
The Protein Multimodal Network is a framework established to understand and explain complicated structures made up of long sequences of amino acids in proteins. These amino acids have complex 3-D structures. PMN acts as an expert detective, putting together diverse evidence regarding a protein’s structure as well as behavior to create a complete picture. The fundamental idea behind PMN is that two amino acids could exist far apart in the linear sequence yet close in 3D space. Using an efficient learning method, PMN describes many ways to understand the proteins into one unified model. It uses a combination of sequential and graph Transformers, which are effective for comprehending complicated relationships. They help to capture essential long-range interactions inside the structure of proteins, which may be thought of as long-range networks.
The model focuses on protein 3D structure as well as amino acid sequence. To analyze and comprehend protein structures, it employs specialized components such as a local encoder, a GVP module, and a global Transformers encoder. As a consequence, a complete representation of a protein is created that effectively collects both local and global information.
Prediction of Ligand Generation and Binding Affinity
In drug development, it is crucial to predict how successfully a potential drug molecule (ligand) will connect to a certain protein structure and to build entirely new drug-like ligands that may bond to that structure. The search begins with a dataset of known protein-ligand pairings, each with a binding score indicating their interaction’s intensity.
Considering the 3D structures of proteins and ligands to predict binding affinity. A molecular graph and a binary fingerprint that incorporates important structural features are used to depict a drug-like ligand. These representations are provided in specialized neural networks, and their embeddings are integrated with the representation of the protein using another neural network. This unified input is then used to forecast a binding affinity score, which indicates how strongly the ligand may bind to the protein.
For ligand generation, rather than generating ligands based on a graph or smiles-based method, SELFIES (SELF-referenced Embedded Strings) are utilized, which ensures molecular validity. A ligand is essentially a string composed of SELFIES tokens from a predefined set. A new ligand is generated by computing independent probability vectors for each position in the ligand string and selecting tokens based on these probabilities.
Traditional methods often generate similar molecules, lacking diversity. To overcome this, this approach from computer vision allows for a more varied generation. By utilizing the decoder from a pretrained unconditional variational auto-encoder, diversity in ligand generation could be enhanced while maintaining their validity. This methodology combines advanced machine learning, molecular representation, and neural networks to predict how well a molecule will bind to a protein and generate a wide array of novel and valid ligands, providing valuable tools for drug discovery.
Binding Affinity Prediction using DAVIS and KIBA
Binding Affinity Prediction was performed to see how the model predicts the affinity between proteins and ligands, a pivotal aspect in medicine discovery. It used two datasets, DAVIS and KIBA, containing information about proteins and ligands and their list strengths. Still, the original datasets demanded the 3D structure details of the protein targets, so it used augmented data with 3D structures obtained through AlphaFold.
Metrics such as mean squared error (MSE), concordance index (CI), and r2m index were used to assess the model’s performance. MSE helps to understand prediction accuracy by evaluating the difference between observed and expected values. The concordance index evaluates the ranking consistency of projected affinities, while the r2m index reflects the model’s predictive strength.
In the experiments, PMN showed significant improvements over baseline methods, especially on the DAVIS dataset. It outperformed competitors on DAVIS and performed comparably well on KIBA. The approach efficiently utilized protein representations and sequences, showcasing a balance between performance and training efficiency compared to other methods like FusionDTA and BiCompDTA, which either demanded substantial computational resources or carefully processed features.
A dataset called KIBA contained information about proteins and ligands. For generating molecules, another dataset was utilized (ZINC 250K), which included about 250,000 drug-like molecules. These molecules were converted to representations from SMILES to SELFIES format. To evaluate the quality of the generated molecules, various metrics like Fréchet ChemNet Distance (FCD), drug-likeness (QED), penalized lipophilicity (pLogP), and synthetic accessibility (SA) were used. FCD helps measure the similarity between generated and real-world molecules. The binding affinity of the generated ligands to specific protein targets was also accessed using docking measurements.
The experimental results demonstrated that TargetVAE could accurately generate molecules that closely resemble real-world molecules, especially when trained using a specific objective (Ofor). These generated molecules showed promising drug-like properties and reasonable binding affinities to target proteins, offering a valuable step toward target-aware drug design.
Trying out the Atom 3D Ligand Binding
The atom 3D Ligand Binding focuses on the Ligand Binding Affinity (LBA) dataset within the Atom 3D benchmark. The LBA dataset provides information about the binding regions on protein surfaces and their complexes with drug-like molecules. The PMN model was modified to operate specifically on the 3D structures of protein-ligand complexes, excluding the language modeling part.
For the evaluation, the modified model was compared with several existing methods, including ENN (Cormorant neural network), 3D CNN, GNN (Graph Neural Network), and GVP-MPNN (Graph Variational Propagation Message Passing Neural Network). The evaluation was based on the root-mean-squared error (RMSE) metric.
The results demonstrated that employing Transformers to model long-range interactions among atoms significantly improved the model’s performance compared to local networks, especially GVP-MPNN, which had similar input features. Moreover, the approach showed efficient computational performance comparable to other methods.
Conclusion
TargetVAE and the Protein Multimodal Network (PMN) are reshaping drug development by rapidly creating putative drug-like compounds with strong binding affinities to a wide range of proteins. TargetVAE uses a variational auto-encoder to quickly anticipate ligand synthesis and binding affinity, addressing existing approaches’ time-consuming and expensive nature. TargetVAE greatly improves the diversity and validity of synthesized compounds by utilizing different protein representations and SELFIES for ligand production, which is typically absent in conventional techniques.
TargetVAE’s performance on the DAVIS and KIBA datasets shows significant gains, notably on the DAVIS dataset, demonstrating its usefulness in using protein representations and sequences. The research further extends the use of PMN to Atom 3D Ligand Binding, emphasizing its better performance and computational efficiency compared to other methods.
Article source: Reference Paper
Important Note: bioRiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Prachi is an enthusiastic M.Tech Biotechnology student with a strong passion for merging technology and biology. This journey has propelled her into the captivating realm of Bioinformatics. She aspires to integrate her engineering prowess with a profound interest in biotechnology, aiming to connect academic and real-world knowledge in the field of Bioinformatics.











{kind=link}