
Scientists discuss the study of 150,119 people’s whole genomes from the UK Biobank, which is a group of high-quality variants that includes 58,707,036 indels and 585,040,410 single-nucleotide polymorphisms, or 7.0 percent of all potential single-nucleotide polymorphisms in humans.
Image Source: https://doi.org/10.1038/s41586-022-04965-x
A thorough and trustworthy characterization of both sequences and phenotypic variation is necessary to gain a thorough understanding of how variations in the human genome’s sequence influence phenotypic diversity.
Large cohorts with abundant phenotypic data have been sequenced using whole-exome or whole-genome methods during the past ten years to get insights into this relationship.
In the given work, the scientists discussed the study of 150,119 whole human genomes from the UK Biobank. This was a group of high-quality variants that included 58,707,036 indels and 585,040,410 single-nucleotide polymorphisms, or 7.0 percent of all potential single-nucleotide polymorphisms in humans.
Through the use of a depletion rank score of windows along the human genome, the substantial collection of variants enabled the researchers to describe selection based on sequence variation within a population.
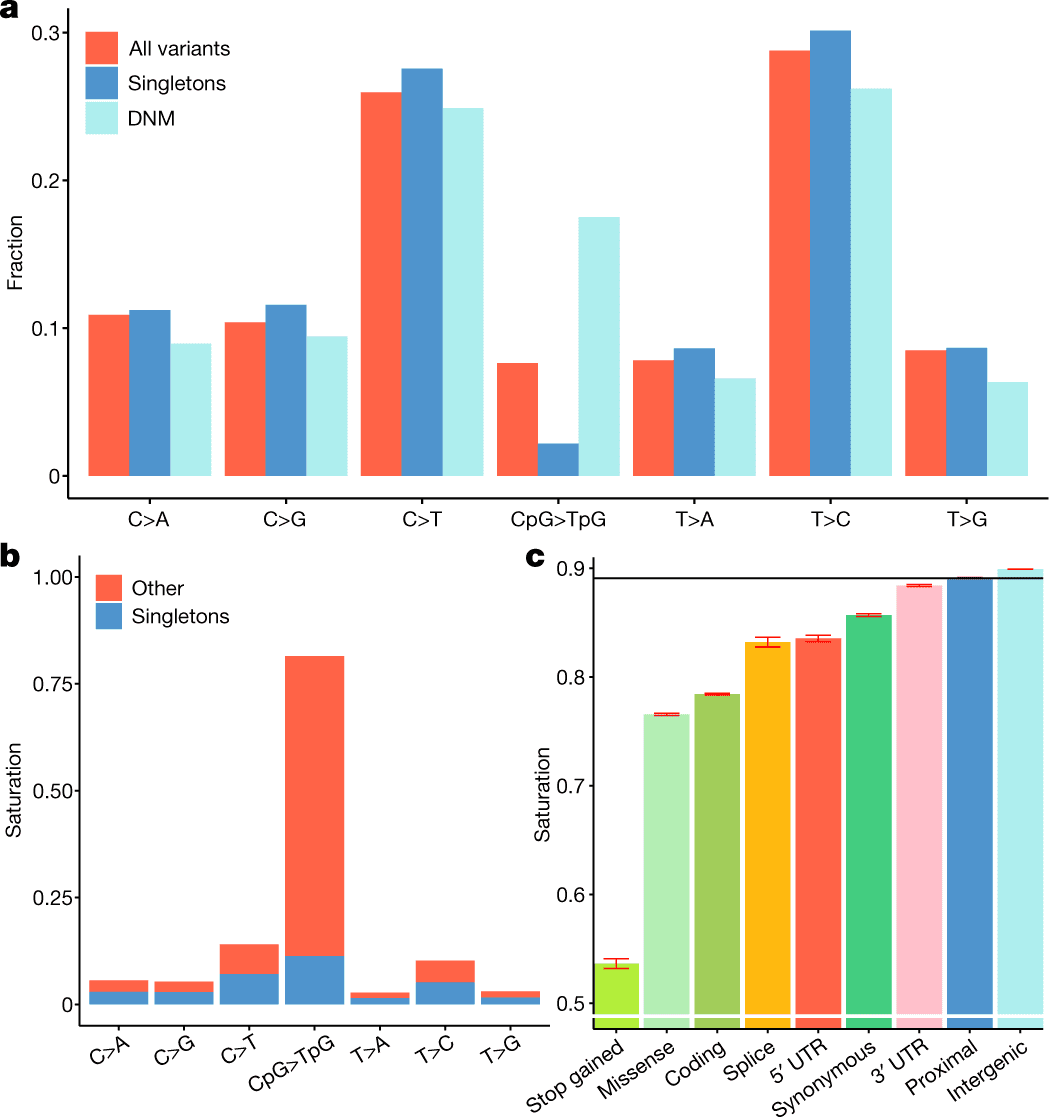
Coding exons are only a small portion of the genome’s areas subject to significant sequence conservation, according to depletion rank analysis.
Within the UK Biobank, they categorized people into three cohorts:
- A sizable British Irish cohort,
- A smaller African cohort, and
- A South Asian cohort.
In order to reliably attribute the majority of variations held by three or more sequenced individuals, a haplotype reference panel was offered.
The scientists found 2,536,688 microsatellites and 895,055 structural variations, which are normally left out of large-scale whole-genome sequencing studies.
They offered many examples of trait connections for rare variants with significant effects that were not previously discovered by research based on whole-exome sequencing and/or imputation using this powerful new resource.
An Unparalleled Opportunity to Study Human Sequence Diversity
With a healthy volunteer bias, the UK Biobank (UKB) documented the phenotypic variation of 500,000 participants throughout the UK.
The UKB whole-genome sequencing (WGS) consortium is sequencing each participant’s entire genome to a depth of at least 23.5 base pairs.
In this article, the scientists presented the first data release based on the WGS of 150,119 individuals, which contains a large collection of sequence variants, including single-nucleotide polymorphisms (SNPs), and short insertions or deletions (indels), microsatellites, and structural variants (SVs).
The joint execution of all variation calls across persons made it possible to compare the outcomes consistently.
The resulting dataset offers a rare chance to research human sequence diversity and how it affects phenotypic variation.
Structural Variation
Whole-exome sequencing (WES) data and genome-wide SNP array data from earlier investigations of the UKB were generated.
Despite the fact that SNP arrays often only capture a tiny portion of the most frequent variants in the genome, imputation can be used to survey a considerably broader collection of variants in these individuals when used in conjunction with a reference panel of WGS individuals.
However, imputation ignores individual-specific variations that are only typed on SNP arrays and yields incorrect results for variants with insufficient haplotype sharing between carriers in the reference and imputation sets.
Poorly imputed variations are frequently caused by structural variation and are typically infrequent, highly changeable, or located in genomic regions with complex haplotype structures.
Functionally Important Variants
Because WES is mostly restricted to regions that are known to be translated, it only detects a small (2–3%) percentage of the sequence variation in the human genome.
Variants outside of coding exons also appear to be functionally significant, explaining a significant portion of the heritability of traits. Variants inside protein-coding regions are relatively easy to assign functions to.
Demonstration from the Study
SNP and short indels have traditionally been the focus of large-scale sequencing studies.
These types of variants are the most prevalent in the human genome, while others, such as SVs and microsatellites, influence more base pairs overall and are therefore more likely to have functional effects.
Due to the capture stage of targeted sequencing, even the SVs that overlap exons are challenging to identify with WES due to the considerably larger variability in the depth of sequence coverage in WES studies than in WGS research.
In extensive sequence analysis investigations, microsatellites—polymorphic tandem repeats of 1-6 bp—are also frequently not studied.
In the study, the scientists highlighted a few discoveries made feasible by this enormous new WGS data collection that would be difficult or impossible to make using WES and SNP array datasets.
Defining of Cohorts
The majority of UKB data investigations to date have focused on a list of 409,554 people who self-identified as “white British” and clustered according to genetic principle components determined from microarray genotypes.
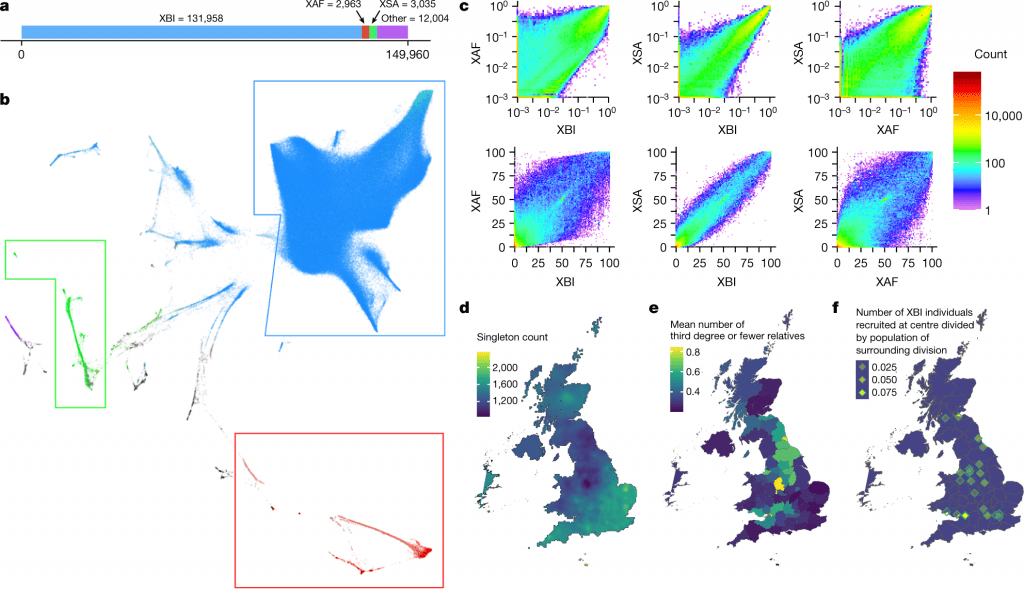
Image Source: https://doi.org/10.1038/s41586-022-04965-x
The scientists aimed to take advantage of the diversity in the UKB, similar to several recent studies.
To do this, they utilized a combination of:
- Uniform manifold approximation and projection (UMAP) dimension reduction of 40 genetic principal components provided by UKB, and
- ADMIXTURE analysis supervised five reference populations and self-reported ethnicity information to define three cohorts based on the most prevalent ancestries identified among the participants.
The Discovery of Rare Non-Coding Sequence Variants
The dataset produced by sequencing the whole genomes of over 150,000 UKB participants is unmatched in size and offers the most thorough analysis of the sequence variation in a single population’s germline genomes to date. The scientists identified a large number of sequences.
With the help of two sets of SNP and indel data, as well as data from microsatellites and SV, two variation classes that are typically not examined in GWAS, they described a large number of sequence variants in the WGS subjects.
When compared to the WES of the same individuals, the number of SNPs and indels is 40-fold higher.
Even inside annotated coding exons, 10.7% of variations discovered by WGS are missed by WES.
Most of the remaining genome, including functionally significant UTRs, promoter regions, and unannotated exons, are not covered by WES.
The identification of uncommon non-coding sequence variants with greater effects on height and menarche than any variants revealed in GWAS to date serves as an illustration of the significance of these regions.
The Endpoint
Although more analysis is required to fully comprehend the DR score’s features, ramifications, and relationship to other measures of conservation and sequence constraint, the researchers anticipated it to be a valuable tool for finding genomic regions of functional importance.
A low DR score indicates that coding exons are subject to strong purifying selection, yet they consist of a very small portion of the areas with this score.
Their understanding of the function and significance of the non-coding genome is expected to be much improved by the large-scale sequencing outlined in the study, as well as the ongoing work to sequence the complete UKB.
These results should significantly increase the understanding of the connection between human genome variation and phenotypic diversity when combined with the thorough characterization of phenotypic diversity in the UKB.
Paper Source: Halldorsson, B.V., Eggertsson, H.P., Moore, K.H.S. et al. The sequences of 150,119 genomes in the UK Biobank. Nature (2022). https://doi.org/10.1038/s41586-022-04965-x
Learn More About Bioinformatics:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}