
The University of Cincinnati scientists introduce iLINCS, an integrative web-based platform for analyzing OMICS data and markers of cellular disturbances. The user-friendly interfaces of iLINCS allow for the execution of sophisticated omics signature analyses, mechanism of action analysis, and signature-driven drug repositioning.
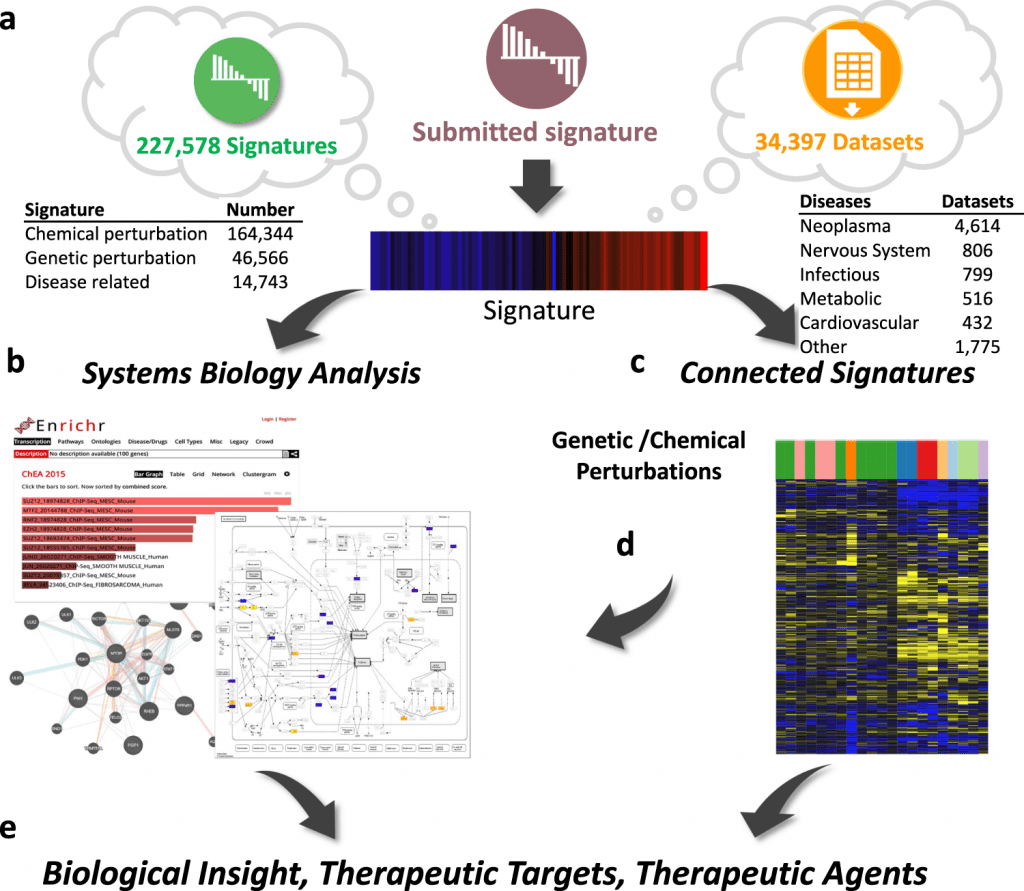
Image Source: https://doi.org/10.1038/s41467-022-32205-3
Only a few systems exist that combine several omics data types, bioinformatics tools, and user interfaces for integrative analysis and visualization without the need for programming knowledge.
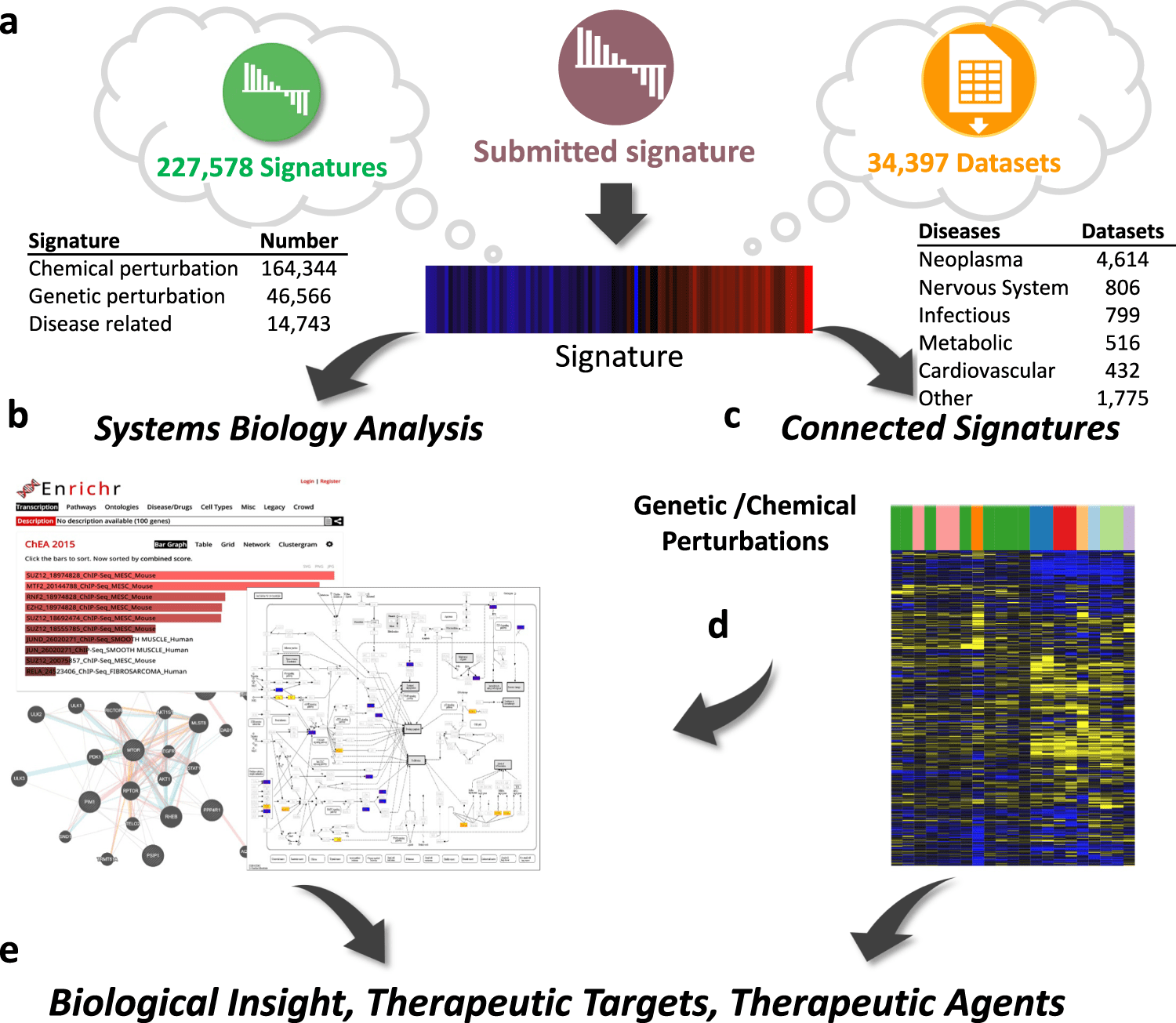
The platform enables the study of user-submitted omics signatures of diseases and cellular perturbations in addition to mining and re-analysis of the enormous collection of omics datasets (>34,000), pre-computed signatures (>200,000), and their relationships.
Large-scale omics data sources, a variety of analytics tools, and interactive visualization tools are all combined in iLINCS analytical processes to create a comprehensive platform for omics signature analysis.
The intuitive user interfaces of iLINCS make it possible to carry out complex omics signature analyses, mechanism of action investigations, and signature-driven drug repositioning.
They employ the study of cancer proteogenomic signatures, COVID 19 transcriptome signatures, and mTOR signaling as three use cases to demonstrate the usefulness of iLINCS.
Omic Signatures of Diseases
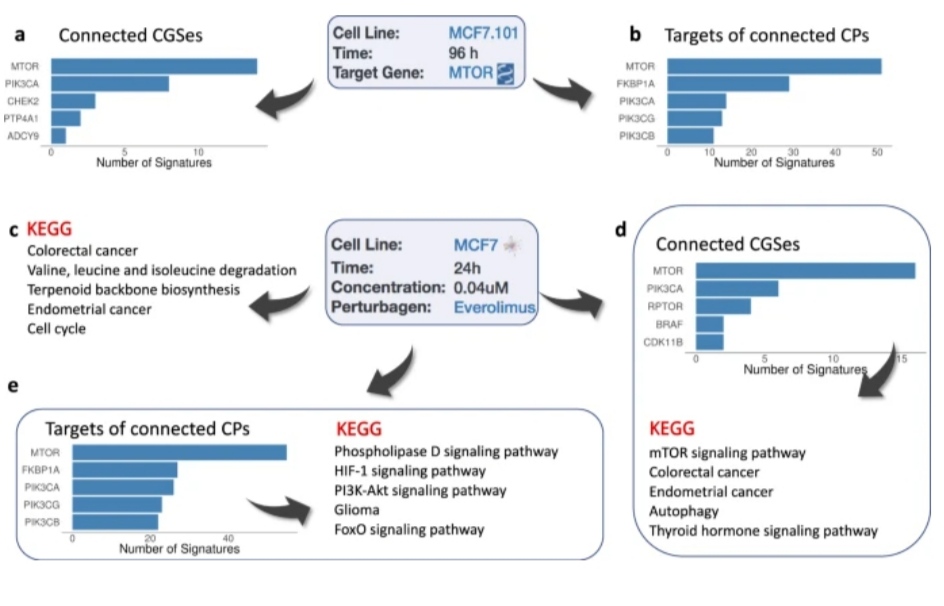
Image Source: https://doi.org/10.1038/s41467-022-32205-3
Changes in gene or protein expression levels following a cellular perturbation make up the transcriptomics and proteomics profiles in response to the disruption.
A high-dimensional readout of cellular state change called an omics signature tells us about the biological processes that are impacted by perturbations and the phenotypic changes that these perturbations cause in the cell.
Even if it isn’t always clear from the signature alone, the molecular processes through which the perturbation results in the observed changes are revealed.
The changes in gene/protein expression levels between diseased and healthy tissue samples constitute the omics hallmark of an illness if we define a disease as a disruption of the homeostatic biological system under normal physiology.
Opening of New Learning Avenues
There are many transcriptomics datasets and signatures available because of the affordability and efficiency of transcriptomics assays.
Large-scale proteomics signature production is now possible thanks to recent developments in high-throughput proteomics. Several recent initiatives have focused on the systematic production of libraries of signatures by re-analyzing public domain omics datasets and the systematic generation of omics signatures of cellular perturbations.
Transcriptomic signatures were produced at a scale never before seen in the library of integrated network-based cellular signatures L1000 dataset.
The accessibility of the resulting libraries of signatures creates fascinating new opportunities for understanding disease causes and the pursuit of efficient treatments.
Chemical Perturbagen Signature Correlation
Numerous studies have been reported on omics signature analysis and interpretation. For identifying changes in molecular phenotypes implicated by transcriptional signatures, a variety of techniques and tools have been created using gene set enrichment, pathway, and network analysis approach.
The Connectivity Map approach to finding prospective therapeutic candidates is based on directly comparing transcriptional patterns of disease with negatively correlated transcriptional signatures of chemical perturbations.
Similar to how possible pharmacological and chemical perturbagen targets have been found, chemical perturbagen signatures have been correlated with genetic alterations of particular genes.
The iLINCS Portal
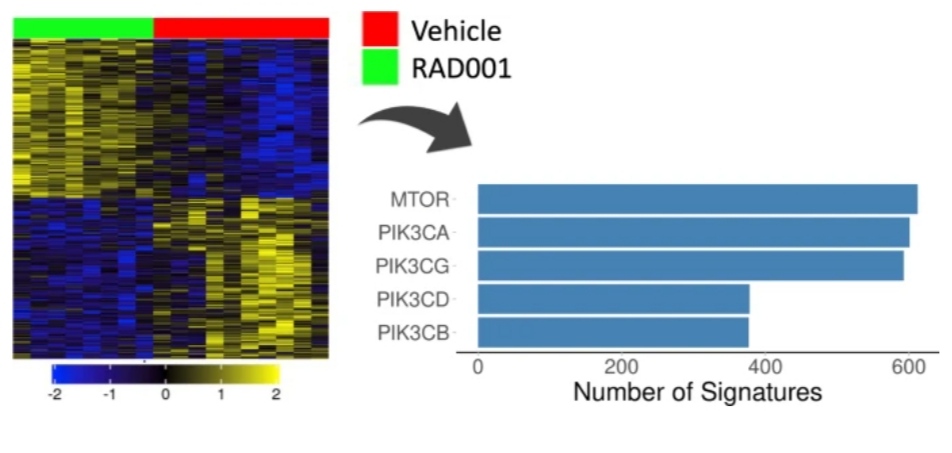
Image Source: https://doi.org/10.1038/s41467-022-32205-3
New user-friendly integrative tools that are accessible to a significant portion of the biomedical research community are required to bring these data together to fully exploit the information present in omics signature libraries and within the countless omics signatures generated frequently and constantly by researchers around the world.
The integrative LINCS (iLINCS) portal creates a robust system for omics signature analysis by combining libraries of pre-computed signatures, structured datasets, and relationships between signatures with a bioinformatics analysis engine and optimized user interfaces.
The Endpoint: iLINCS for the Integration of Public Domain Data and Signatures with a User-Friendly Analysis Toolbox
A distinctive integrated platform for the study of omics signatures is iLINCS. The extensive array of studies made possible by the interconnected analytical workflows and the huge quantities of omics datasets, signatures, and their linkages are only partially covered by the few canonical use cases that are discussed here.
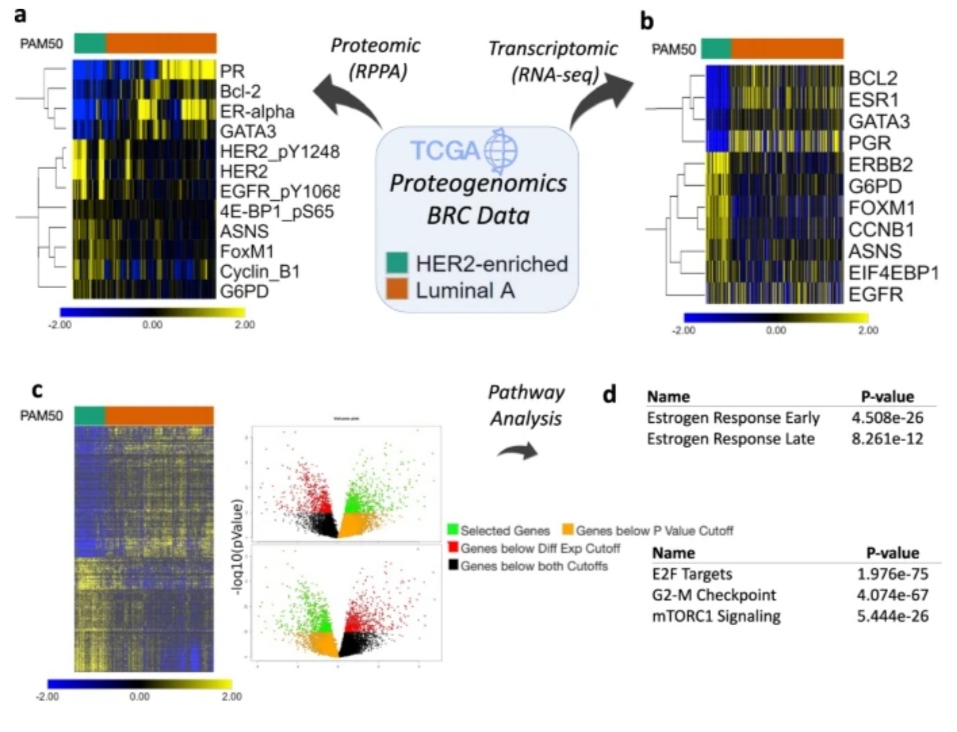
Image Source: https://doi.org/10.1038/s41467-022-32205-3
The iLINCS GUI was only accessed by using the mouse to complete all presented use cases. As seen in the online documentation and video demonstrations, each use case may be finished in about 5 minutes.
iLINCS has been employed in a variety of methods in the published research so far, and a variety of disorders have been studied.
iLINCS enables creative procedures for biological interpretation of omics signatures using CMAP analysis in addition to supporting traditional analyses.
In Use Case 1, the scientists demonstrate how CMAP analysis in conjunction with pathway and gene set enrichment analysis can reveal a chemical perturbagen’s mode of action when standard enrichment analysis carried out on the differentially expressed genes is unable to identify specific signaling pathways.
Similar to this, variations in the proteome profiles of the neurons from schizophrenia patients have been linked, first with the LINCS CGSes of the respective genes and then with LINCS CP signatures, to discover possible treatment drugs.
These investigations allowed for the discovery of PPAR agonists as potential therapeutic drugs with the potential to reverse the bioenergetic signature of schizophrenia, which were later shown to affect behavioral traits in a rat model of schizophrenia.
Future extensions might be easily incorporated into the iLINCS platform because it was designed to be flexible.
There are countless chances to develop extra analytical workflows thanks to the improved database representation and R analysis engine.
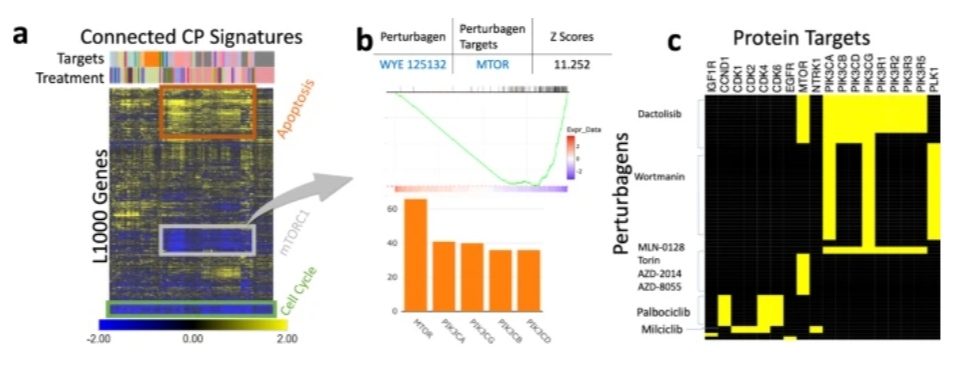
Image Source: https://doi.org/10.1038/s41467-022-32205-3
Adding information to backend databases allows collections of omics datasets and signatures to be expanded. The creation of workflows for the fully integrated analysis of many datasets and various omics data types will be one of the key areas for expanding iLINCS functionality.
When it comes to the integrative analysis of matched transcriptomic and proteomic data, iLINCS makes it possible to integrate data so that the genes and proteins studied in one omics dataset are informed by the findings of the other dataset in Use case 2.
However, more useful signatures may be produced by performing a direct integrative analysis on both types of data.
The breadth of such integrated analyses will also be expanded by the incorporation of additional proteomics datasets, such as the Clinical Proteomic Tumor Analysis Consortium (CPTAC) collection.
There are numerous, molecularly different subtypes of many complicated illnesses, including cancer. Building efficient disease signatures for CMAP analysis requires taking these variances into account.
In Use Case 2, the scientists show how to apply iLINCS to compare molecular subtypes when the subtype information is present in sample metadata. A future version of iLINCS will enable the de novo construction of molecular subtypes by cluster analysis, as is frequently done when analyzing cancer samples.
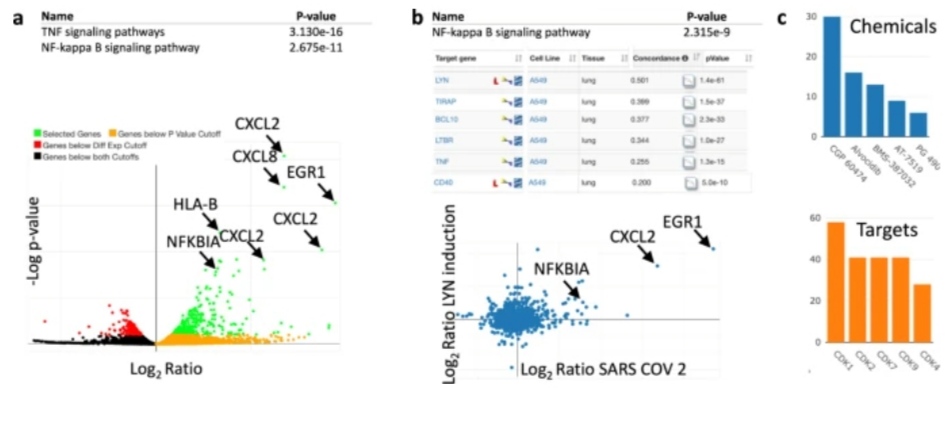
Image Source: https://doi.org/10.1038/s41467-022-32205-3
The procedure for creating disease signatures from single cell datasets is a future expansion that is currently under development.
Numerous single-cell RNA-seq (scRNA-seq) datasets are present in iLINCS, although their analysis is currently carried out using the same methods as the bulk RNA-seq data.
A specialized workflow will produce more accurate disease signatures and more potent CMAP analysis when used to extract disease signatures from scRNA-seq data.
In order to avoid false positive results and identify the most important affected pathways, one must carefully examine the results of CMAP-based pathway analyses.
This is because a lot of signatures are used in CMAP analysis, and a lot of genes are perturbed by either genetic or chemical perturbations. In the signature-similarity-based pathway analysis, it is crucial to be aware of the limitations of conventional gene enrichment pathway analysis linked to overlapping pathways.
Additionally, the hierarchical architecture of gene expression regulation may cause the perturbation of genes at various levels of the regulatory processes to produce transcriptional signatures that are similar to one another (e.g., signaling proteins vs. transcriptional factors). Changes in specific signaling pathways affect how quickly cancer cell lines divide, and it is anticipated that the ensuing transcriptional profiles will have some similarities in terms of the up and down-regulation of proliferation drivers and indicators.
Simultaneously, it is anticipated that signatures corresponding to changes in proteins that control the same groups of biological processes will show a higher degree of similarity.
The “proliferation” component is included in each of the top 100 chemical perturbagen signatures that are adversely linked with the Her2E breast cancer signature in Use case 2.
However, a portion of the most strongly connected signatures is more explicitly linked to mTOR inhibition, suggesting that mTOR signaling modulation influences proliferation to some extent.
Despite being true, the connection to disturbances that affect cellular proliferation might potentially be unreliable since it is so general.
Different proliferation rates can be explained by a higher-level molecular explanation thanks to the link with mTOR signaling, which is more specific.
For reviewing these fine details in Use case 2 and for adequately interpreting the findings of a CMAP analysis, iLINCS offers methods for closely examining gene expression patterns in signatures discovered by CMAP analysis.
For the examination and mining of LINCS L1000 signature libraries, a number of online tools have been developed. They enable the creation of automated workflows for in-depth study of transcriptomics data and signatures as well as online searches of L1000 signatures.
The Broad Connection Map team uses the clue.io query tool, which was created by the LINCS Transcriptomic Center at the Broad Institute, to analyze connectivity in user-submitted signatures.
Since iLINCS replicates the connectivity analysis capabilities, similar queries between the two systems may yield qualitatively similar answers.
However, iLINCS has a far wider focus. Additionally to Connection Map datasets, it offers connectivity analysis using signatures and offers a wide range of primary omics datasets so that users can create their own signatures.
Additionally, iLINCS’ analytical methods support deep systems biology research and knowledge discovery for both omics signatures and the genes and protein targets discovered through connectivity analysis.
For users without a background in programming, iLINCS reduces technical barriers to the reuse of publicly accessible omics datasets and signatures.
With a basic grasp of omics data analysis, most scientists should be able to use the user interfaces without any help. Public domain omics data are becoming easier to find and reuse because of recent work in standardizing and indexing.
iLINCS is merging public domain data and signatures with a user-friendly analytical toolset, which is the next natural step.
Additionally, the iLINCS GUI’s analysis phases are all powered by an API that can also be used to power the functionality of other web analytic tools and computational pipelines based on scripting languages, including R, Python, and JavaScript.
This makes iLINCS a natural tool for data scientists who like point-and-click GUIs and scientists who want to analyze omics signatures using scripted analytical pipelines.
Article Source: Pilarczyk, M., Fazel-Najafabadi, M., Kouril, M. et al. Connecting omics signatures and revealing biological mechanisms with iLINCS. Nat Commun 13, 4678 (2022). https://doi.org/10.1038/s41467-022-32205-3
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}