
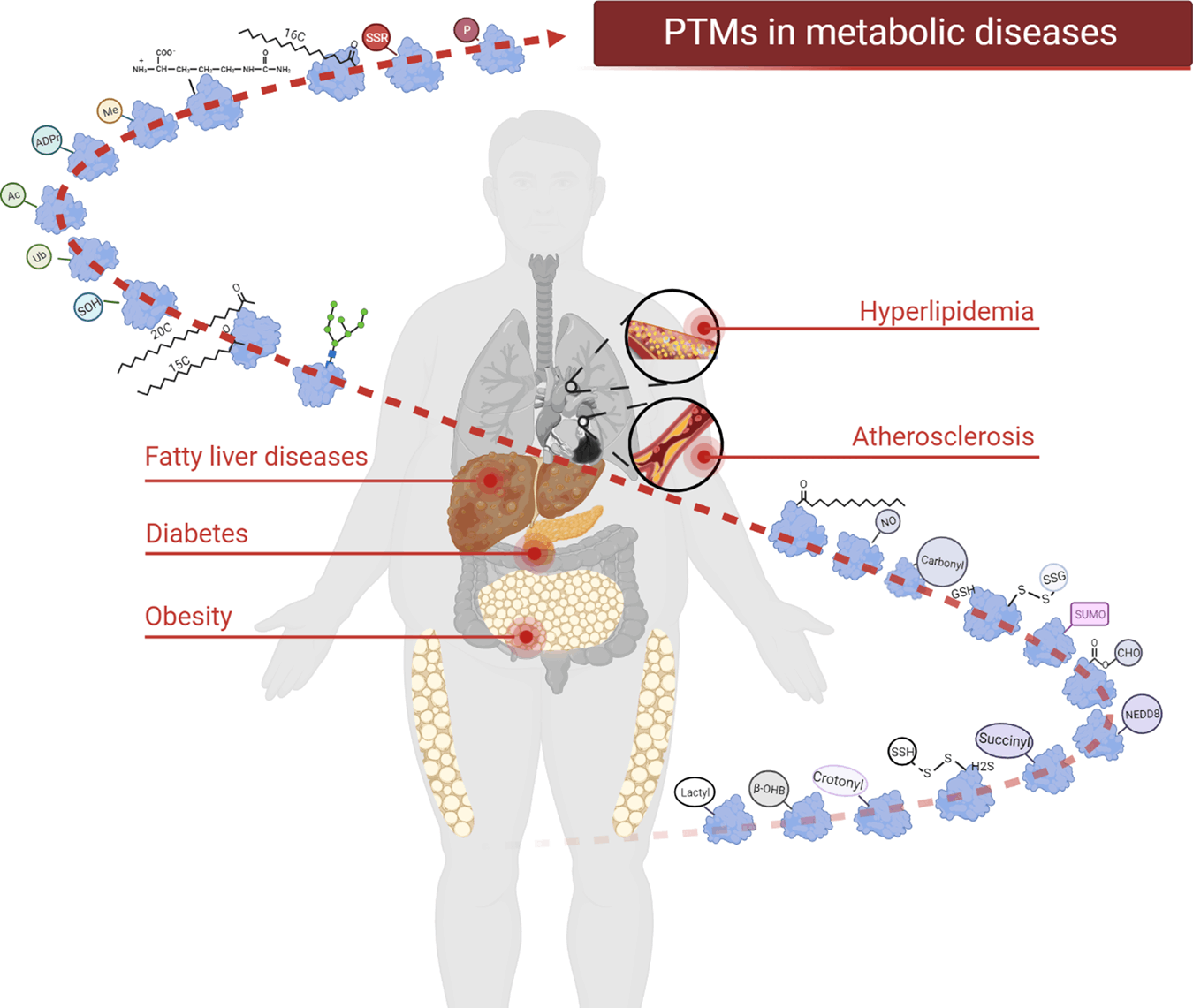
Comprehensive introspection of cellular and molecular level aberrations that prompted the worldwide prevalence of life-threatening Non Communicable Diseases like Diabetes, Obesity, Hypertension, Nonalcoholic Fatty Liver Disease (NAFLD), CDV (Cardiovascular diseases), etc. is an acute need towards their prohibition, effective clinical treatment, control, and remedy. Decoding the intricate details, cascade, and consequences of protein post translational modifications hold promising prospects in this aspect.
Introduction to Protein Modification
Despite having a limited number of genes in the genome, Alternative splicing and Post Translational Modifications (PTMs) contribute to a multifarious proteome consisting of approximately 1 million Proteins and their remarkably dynamic life-sustaining functions. Post Translational Modifications refer to the chemical modification of amino acid side chain in proteins after or during their biosynthesis. PTMs reversibly or irreversible alter protein structures, influence and diversify function, activation, assembly, interactions, stability, folding, turnover, and localization of proteins and thus facilitate protein cross talks that directly contribute to maintaining overall cell physiology through fine-tuning molecular trafficking, cell metabolism, signal response pathways, intracellular & extracellular communication, gene expression, DNA repair, and Cell cycle. PTMs can occur at any stage of a protein’s ‘lifespan’ in the nucleus, cytoplasm, Golgi complex, and Endoplasmic Reticulum.
Post Translational Modification (PTM) Variants
Post translational modifications recognition remained stagnant for a long owing to a lack of technology. The enrichment in genomic data acquisition and amelioration of detection approaches such as immunoprecipitation, mass spectrometry (MS)-based techniques, radioactive isotope labeling, immunofluorescence techniques, peptide/protein array, and proximity ligation assay (PLA) assisted in the progress of PTM research. Over 600 types of PTMs have been listed in the UniProt database.
PTMs and Diseases
The interplay of PTMs is very tightly regulated, but lifestyle or genetic factors can create an imbalance in their homeostasis. It manifests as a diverse range of diseases. A brief overview of PTM’s, along with their implication in Metabolic disease progression, is provided below. Note that aberrations in PTM can generate innumerable disorders which aren’t mentioned here. Let us now look at several noteworthy post-translational modifications that play a crucial role in the pathogenesis of metabolic diseases.
- Phosphorylation
Phosphorylation is the kinase enzyme-mediated linkage of a Phosphate group from ATP (Adenosine Triphosphate) to amino acids, specifically Serine, Threonine, and Tyrosine residues of a protein. This process can be reversed by Phosphatases via dephosphorylation. This phenomenon supervises cellular processes relevant to environmental responsiveness, immune response, replication, transcription, apoptosis, and cell metabolism. Phosphorylation can rapidly control the function of proteins through two mechanisms: activate enzymes through allosterism or enable interaction of domains to activate signal transduction.
Insulin secretion from islets of Langerhans and Insulin-mediated signaling responses both are associated with Phosphorylation events and kinases. Based on the SILAC method, 8539 phosphosites derived from 2487 proteins were identified in the islets, and 170 phosphosites were differentially expressed in response to a short-term high glucose spike. In Type 2 Diabetes (T2D), there is increased phosphorylation at threonine 213 and tyrosine 361 sites of ATPsyn-β.
STAT3 phosphorylation deficiency in the hypothalamus renders central leptin-induced obesity. Protein kinase (PKs) families act on multiple downstream key protein targets in NAFLD and regulate hepatic gluconeogenesis, lipogenesis, and inflammation. MAPKs (Mitogen Activated Protein Kinase) are linked to atherosclerosis via matrix production and foam cell formation, VSMC proliferation, and migration.
- Acetylation
Acetyltransferases facilitate chemical modification by adding an acetyl group (-COCH3) derived from acetyl coenzyme A (acetyl-CoA) to the ε-carbon of lysine, and this reaction is reversed by deacetylases that exclude the acetyl group from peptide slide chain. Moreover, acetylation can be regulated by acetyl-CoA using nonenzymatic regulation. The dynamic balance between histone acetylation and deacetylation in the nucleus adjusts gene expression.
A High Fat Diet (HFD) can trigger the process of acetylation in β-hydroxylacyl coenzyme A dehydrogenase and acyl-CoA dehydrogenase (LCAD), ultimately interfering with the insulin signaling pathway.
Inhibition of HDAC6 (Histone Deacetylase) in pancreatic islets downregulates insulin signaling. Upregulated HDAC7 impairs insulin secretion and contributes to β cell dysfunction in T2D islets.
Protein acetylation, energy metabolism, and adiposity are conjoint. The H3 (Histone) acetylation at lysine 9 & 18 sites of Tnfα and Ccl2 genes are found upregulated in obese mouse livers.
Protein acetylation can regulate metabolism in chronic liver diseases. Hyperacetylated-LDHB (Lactate Dehydrogenase-B) has been detected in NAFLD samples. HDAC9 is known for its role in regulating atherosclerotic aortic calcification.
- Methylation
During Methylation, methyl groups are transferred from active methyl compounds to amino acid residues. Methylation is associated with fine-tuning transcriptional regulation and epigenetic silencing.
The histone methyltransferase SETDB2-associated pathway IFN-β-SETDB2-H3K9me3 is dysfunctional in diabetes and induces NFκB-mediated inflammation. Deficiency of PRMT5 (Protein arginine methyltransferase 5) hampers glucose-stimulated insulin secretion in a mouse model.
H2A E67me1 and H4 E74me1 are considered to be associated with the pathological process of obesity. Histone 3 lysine 9 methyltransferase enzyme (G9a) is downregulated in diet-induced animal models of obesity.
The early stages of NAFLD mark a notable reduction in the activity of glycine N-methyltransferase (Gnmt). Histone methylation profile in human atherosclerotic lesions revealed the global downregulation of H3K27me2 and H3K9me2 in atherosclerotic plaques. Histone methyltransferases and a few intermediate products during the enzymatic process are implicated in atherosclerotic progression.
- Ubiquitination
Ubiquitin is a highly conserved polypeptide of 76 amino acids. Three steps are involved in ubiquitination: ubiquitin activation, conjugation, and ligation. Ubiquitin-proteasome system degrades proteins and recycles amino acids. Ubiquitination is regulated by the conjugation of ubiquitin by ubiquitin ligases, and deubiquitinating enzymes remove ubiquitin. The maintenance and development of stem cells, as well as other cellular functions, including DNA replication, transcription, autophagy, apoptosis, innate immunity, and signal transmission, depend on ubiquitination.
UBC9 (Ubiquitin Conjugating Enzyme) is downregulated in muscle tissues of T2DM patients. TRIM family proteins are involved in the progression & complications of diabetes. Ubiquitination via E3 ubiquitin ligase and proteasome-dependent degradation of PPARγ (Peroxisome Proliferator-Activated Receptor gamma) exhibit mechanisms in obesity inception. Ubiquitin-specific peptidase 10 (USP10) decreases over time in patients with NAFLD and HFD mice. E3 ubiquitin ligases are recognized to play roles in atherosclerotic inflammatory and metabolic processes.
- SUMOylation
In SUMOylation 10-kDa polypeptide links to the ɛ-amino groups of lysine residues via isopeptide bonds. It’s a highly dynamic enzymatic cascade constituting SUMO-activating E1 enzyme (SAE1/UBA2), SUMO-conjugating E2 enzyme (UBC9), and SUMO E3 ligase and SENPs responsible for deSUMOylation. SUMOylation can occur in cell nuclei, cytoplasm, plasma membrane, ER, and mitochondria.
T2D patients with severe insulin resistance have lower UBC9 protein expression in skeletal muscle. Mice depleted of the UBC9 in pancreatic beta cells spontaneously develop diabetes because of β cell death occurring due to reactive oxygen species accumulation.
SUMO E2 enzyme UBC9 and SUMO E3 ligase PIASγ induce dyslipidaemia by SUMOylating PPARα and suppressing PPARα transcriptional activity. Dyslipidemia in atherosclerosis is a crucial metabolic risk factor.
- Glycosylation
Glycosyltransferase or Glycosidase links oligosaccharide chains to specific residues by covalent bonds. Glycosylation takes place in the endoplasmic reticulum, Golgi apparatus, cytosol, and sarcolemma membrane. N-glycosylation and O-glycosylation are important in the supervision of protein conformation and function.
In Maturity-onset diabetes of the young (MODY), fucosylated N-glycans are a novel biomarker. O-GlcNAc modification can be a potential biomarker and assist in identifying prediabetic patients. Increased AGEs (advanced glycosylation end products) promote oxidative stress and contribute to cardiovascular diseases in diabetic patients.
Habitual HFD consumption can increase O-glycosylation levels in cerebral arteries and the heart. Recently, overexpression of glycosyltransferase 8 domain containing 2 (Glt8D2) was noticed in patients with severe NAFLD. The galNAc-T2 enzyme catalyzes the GalNAc linkage in O-glycosylation. The GALNT2 variant has been shown to affect HDL and triglyceride levels in human genetics studies.
- Neddylation
In Neddylation, ubiquitin-like protein NEDD8 is conjugated to its target proteins. More recently, neddylation has been found to be deregulated in patients with liver fibrosis. The de-neddylation enzyme COP9 signalosome 5 (CSN5) was proposed to inhibit atherosclerosis by regulating macrophage activation. Therefore, de-neddylation approaches may serve as a potential candidate against atherogenesis.
- Palmitoylation
Protein palmitoylation is defined as the process by which palmitic acid (a sixteen-carbon saturated fatty acid) reversibly attaches to cysteine residues via thioester bonds. This increases protein-lipid bilayer interaction. The mechanism is essential for allowing proteins to shuttle between organelles and the membrane.
Palmitoylation has been implicated in the metabolic dysregulation of β-cells. A palm oil-rich diet (HPD) provokes obesity by rendering S-palmitoylation. The latest research detected the upregulated palmitoylation of FAT/CD36 in NAFLD.
- Myristoylation
During myristoylation, the fourteen-carbon saturated fatty acid, myristic acid, attaches to the N-terminal of glycine. Dysregulation in myristoylation has been reported in the onset of cancer, neurological disease, and metabolic disorders.
- Prenylation
Prenylation describes the isoprenoid’s addition to the carboxyl-terminal or cysteine residues. Three isoprenyl transferases catalyze these post translational modifications. Prenylation includes farnesylation and geranylgeranylation. Perturbation in Prenylation is noted in the development of tumors, neurodegenerative and cardiometabolic diseases.
- Glutathionylation
S-glutathionylation describes the reversible bond formation between glutathione (GSH) and the thiol group (-SH) of cysteines. This process regulates insulin resistance in diabetes. S-glutathionylation of hemoglobin in diabetic and hyperlipidemic patients was found to be increased. Serum S-glutathionylated protein levels are elevated in patients with atherosclerosis. S-glutathionylated ApoB100 (an atherogenic lipoprotein) positively influences peripheral vascular damage.
- S-nitrosylation
S-nitrosylation is the covalent incorporation between the nitrosyl moiety of NO and target molecules. S-nitrosylation occurs at the cysteine thiol group, producing protein S-nitrosothiols (SNOs). It is involved in insulin resistance by inactivating Akt. Nitric oxide (NO) inhibits the Abeta-degrading activities of insulin-degrading enzyme (IDE) through S-nitrosylation. Hyper-nitrosylation of RyR2 in β-cells impairs GSIS and blood glucose clearance. S-nitrosylation (SNO) can promote the conversion of NAFLD to NASH (Nonalcoholic steatohepatitis) via the peroxisome PPARγ/SFRP5 pathway. S-nitrosylation inhibits the HSP90-ATPase activity 1 (AHA1) interaction but stimulates the HSP90-cell division cycle 37 (CDC37) association, which modulates endothelial dysfunction and exacerbates atherosclerosis.
- Sulfhydration
Alteration of the thiol group (-SH) in cysteine residues to a persulfide (-SSH) group is Sulfhydration. Mechanically, H2S facilitates its role through protein sulfhydration. H2S is a novel factor mediating obesity and associated metabolic diseases. Plasma H2S levels were proved to be reduced in overweight participants. The deficiency of CSE (cystathionine-γ-lyase) in the liver stimulates the pathogenicity of NASH.
- Citrullination
Citrullination describes the deimination process in which arginine irreversibly hydrolyzes to uncharged citrulline. Citrullination caused by inflammation occurs almost exclusively in the pancreas and can be considered a biomarker of beta cell dysfunction in T1D. Several citrullinated proteins have been identified in human coronary artery plaques.
- ADP Ribosylation
ADP-ribosylation is the transfer of ADP-ribose (ADPr) from NAD+ to the target protein and releases nicotinamide (Nam). ADP-ribosylation is a reversible event where ADP-ribosyltransferases (“writers”) covalently add ADPr, whereas ADP-ribosylglycohydrolases (“erasers”) remove ADPr. ADP ribosylation was mediated by the PARP family of enzymes.
PARP was demonstrated to be a pathogenic marker of diabetes and diabetic complications. Hyperglycemia increases PARP activation in diabetic patients. Suppression of PARP1 reduces plasma triglyceride, cholesterol, and LDL levels and enhances HDL levels in HFD-fed mice.
- Carbonylation
Carbonylation is an oxidation process where Reactive Oxygen Species (ROS) can install carbonyl groups in protein side chains. It is a nonenzymatic and deleterious oxidative damage. During the onset of obesity, levels of ROS in adipocytes increase, which covalently modifies the histidine, cysteine, and lysine residues via protein carbonylation.
Conclusion
The knowledge about tackling mismanaged PTM is still at a superficial level. Nonetheless, PTMs can be designated as biomarkers for early disease diagnosis and treatments. Investigations of PTMs can be a boon for the healthcare system via developing and repurposing of drugs. The study of lifestyle improvement to prevent the onset of such diseases is another promising prospect. Lastly, two individuals with the same symptoms might have different underlying causes, so PTM recognition is another important step toward precision medicine.
Article Source: Reference Paper
Learn More:




Aditi is a consulting scientific writing intern at CBIRT, specializing in explaining interdisciplinary and intricate topics. As a student pursuing an Integrated PG in Biotechnology, she is driven by a deep passion for experiencing multidisciplinary research fields. Aditi is particularly fond of the dynamism, potential, and integrative facets of her major. Through her articles, she aspires to decipher and articulate current studies and innovations in the Bioinformatics domain, aiming to captivate the minds and hearts of readers with her insightful perspectives.





.){kind=link}