
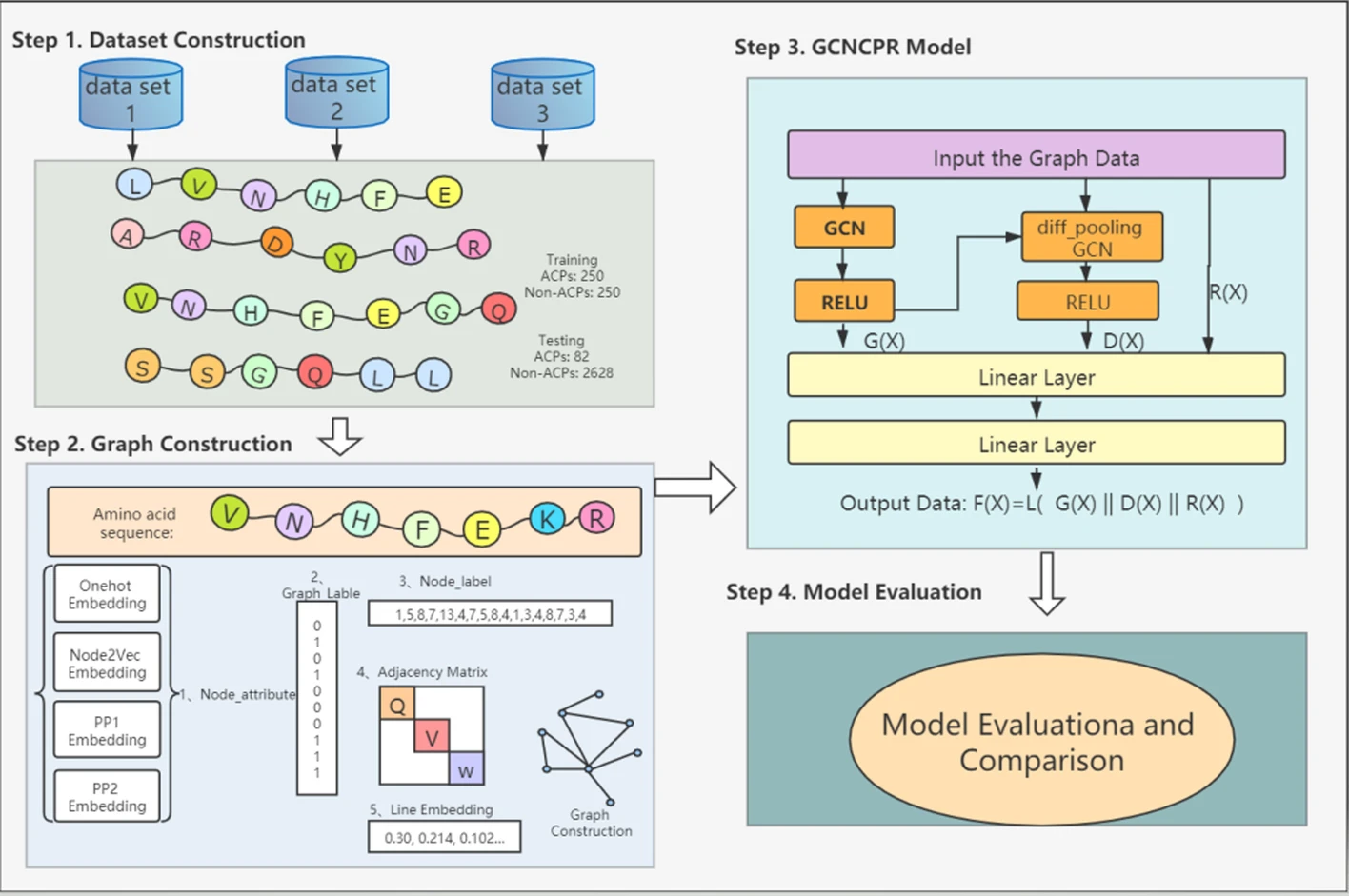
Researchers at the School of Informatics, Xiamen University, China, and Computational Health Informatics Program, Boston Children’s Hospital, USA developed the GCNCPR-ACPs method to effectively capture different levels of node attributes for amino acid node representation learning. GCNCPR-ACPs (a Graph Convolution Neural Network Method is a new machine learning-based method based on collapse pooling and residual network to predict the anticancer peptides), which automatically and accurately predicts anticancer peptides (ACPs) using residual graph convolution networks, differentiable graph pooling, and features extracted using peptide sequence information extraction.
An anticancer peptide inhibits and kills tumor cells. Research on anticancer peptides is of great significance for developing new anticancer drugs, and the prediction of anticancer peptides and non-anticancer peptides is the new hotspot.
ACPs: one of the most effective anticancer medicines
It is disturbing how many individuals globally pass away from cancer each year. Traditional chemotherapy is the main cancer treatment approach. Chemotherapeutic anticancer medications can successfully cure cancer and destroy cancer cells, but they can also harm healthy cells and lead to cancer cell resistance. Therefore, there is an urgent requirement for more rational and efficient treatment medications. Small molecular peptides known as anti-microbial peptides (AMPs) are created by organisms and have the ability to destroy certain cancer cells.
Anticancer peptides are a subset of anti-microbial peptides with anticancer potential. By controlling gene expression and stimulating the immune system, ACPs can effectively limit tumor development and destroy cancer cells. Anticancer peptides are one of the most effective anticancer medicines because they are able to bypass the drawbacks of conventional cancer treatment approaches and kill cancer cells without hurting healthy cells. It has been suggested to use certain experimental techniques to ascertain whether a protein has anticancer potential. Wet-lab studies have a number of drawbacks, but their biggest drawback is that they are difficult, expensive, and time-consuming to do. In contrast, as machine learning and deep learning technologies have advanced, in silico predictions of ACPs and non-ACPs have the benefits of being more accurate, faster, and less expensive. A rising variety of forecasting techniques have been put forth to direct the experimental selection of potential ACPs.
In silico predictions of ACP
In the realm of bioinformatics and computational biology, the use of machine learning and deep learning models to distinguish between ACPs and non-ACPs has recently attracted a lot of attention. Experimentally confirmed anticancer peptides are being found and studied increasingly often. The majority of earlier computational techniques employ the already-existing databases to extract the features, and then they apply the feature training model to automatically categorize the peptides into ACPs and non-ACPs.
Image Source: https://doi.org/10.1186/s12859-022-04771-2
Their forecast accuracy has improved as more and more ACP-predicting techniques are developed. The use of diverse computational technologies in cancer therapy will advance quickly thanks to their better prediction power. An enhanced deep learning model called a graph neural network (GNN) has been used for a number of bioinformatics applications, including link prediction, node categorization, and community discovery. There are a growing number of GNN algorithms that might be applied to the anticancer peptides data. Sequence-based characteristics made up the majority of the ACP data, which were then combined with the other features as classifier inputs to create the prediction model. While neglecting the ACP structure and the interaction between amino acids, the majority of conventional classification algorithms for ACP prediction primarily view ACP data as ordered sequence data. The amino acids would be treated as nodes and the connections between amino acids as edges if the ACP data were thought of as a type of structured data, most likely graph data. Then, handling ACP data using graph-based techniques can be made convenient. The node properties of the nodes in the network will also be described using physicochemical information, transforming the ACPs into a topological map made up of amino acid sequences.
The graph-based data of ACPs were handled in the current study using a graph convolution network (GCN) approach based on graph collapse pooling and residual network for predicting ACPs (GCNCPR-ACPs). The graph collapse pooling operator is calculated using the graph convolutional neural network, which also extracts the graph structural characteristics of amino acids. Several nodes are combined into one giant node using the graph collapse pooling module. The peptide chain graph made up of amino acids is ultimately compressed into a big node after going through numerous stages of collapse, and the characteristic of the collapsed large node is the feature of the whole ACP line. The gradient disappearance issue brought on by layer depending is resolved using the residual network.
Primary contributions of the present study:
- The authors introduced a new GCN-based framework called GCNCPR-ACPs for anticancer peptides prediction, which is the first time graph collapse pooling has been adapted for this purpose.
- This is the first attempt to use four types of node attributes for anticancer peptides prediction, consisting of two physicochemical properties of amino acids and two others extracted by one-hot embedding and node2vec methods separately. This is done in order to efficiently capture various levels of node attributes for amino acid node representation learning.
- Using numerous node features, a variety of ACP graph data qualities, and a unique GCNCPR model, the GCNCPR-ACPs technique predicts the ACPs.
- The performance of graph convolutional neural networks that use collapse pooling and residual networks has been thoroughly evaluated through experiments.
Assessing the performance of the GCNCPR-ACPs technique predicts the ACPs
Using these assessment indicators, the performance of the proposed GCNCPR-ACPs model was assessed, and the findings were reviewed. Several machine learning metrics are often employed in prediction systems for performance evaluation.
Ten-fold cross-validation results evaluated the performance of the proposed GCNCPR-ACPs against that of a number of other current predictors, including iACP, ACPred-FL, PEPred-Suite, ACPred-Fuse, AntiCP ACC, AntiCP DC, and Hajisharifi’s, in order to validate its predictive performance.
Results of an impartial test contrasted the performance of the suggested GCNCPR-ACPs with that of numerous other predictors in order to confirm the proposed GCNCPR-ACPs’ robustness. The learning rate, the number of graph convolution network layers, and the assigned ratio are a few crucial factors that affect how well the models function.
Summarizing views on GCNCPR-ACPs
This study suggested GCNCPR-ACPs, a brand-new prediction model. Graph collapse pooling, residual network models, and GCN are effective bioinformatics tools for predicting anticancer peptides. The benefit of the model is its ability to efficiently build the anticancer peptide map. It can extract valuable aspects from graph data, such as adjacency matrices, node labels, line labels, icon labels, and node properties. The innovative model primarily consists of the following modules: the stacked graph convolution neural network module G(X), the graph differentiable pool module D(X), and the residual network module R(X). The suggested predictor can more accurately categorize ACPs and non-ACPs, according to the experimental findings of ten-fold cross-validation and independent testing. The model’s ability to forecast outcomes accurately will hasten its use in the treatment of cancer.
Article Source: Paper Reference
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Riya Vishwakarma is a consulting content writing intern at CBIRT. Currently, she's pursuing a Master's in Biotechnology from Govt. VYT PG Autonomous College, Chhattisgarh. With a steep inclination towards research, she is techno-savvy with a sound interest in content writing and digital handling. She has dedicated three years as a writer and gained experience in literary writing as well as counting many such years ahead.





{kind=link}