
Researchers have introduced a groundbreaking computational framework called DEMINING, designed to directly identify expressed DNA and RNA mutations in RNA deep sequencing data. This innovative approach incorporates a deep learning model named DeepDDR, which effectively distinguishes expressed DNA mutations from RNA mutations following RNA-seq read mapping and pileup. Upon application to RNA-seq data from acute myeloid leukemia patients, DEMINING revealed previously overlooked DNA and RNA mutations, some of which were linked to the increased expression of host genes or the generation of neoantigens. Additionally, the study demonstrated DEMINING’s ability to accurately classify DNA and RNA mutations in RNA-seq data from non-primate species using transfer learning techniques. This development holds great promise for advancing our understanding of genetic mutations and their implications in various biological contexts.
Changes in DNA and RNA play significant roles in the domains of genetics evolution and human well-being. These changes determine the hereditary variety, considering species’ variation and development over the long run. Changes can create genetic issues and illness if they happen in basic and administrative domains. Cancerous growth can also usually be caused by DNA alterations in which oncogenes are activated or deactivated cancer silencer properties. These changes are critical in biodiversity and natural specializations, allowing species to thrive and evolve.
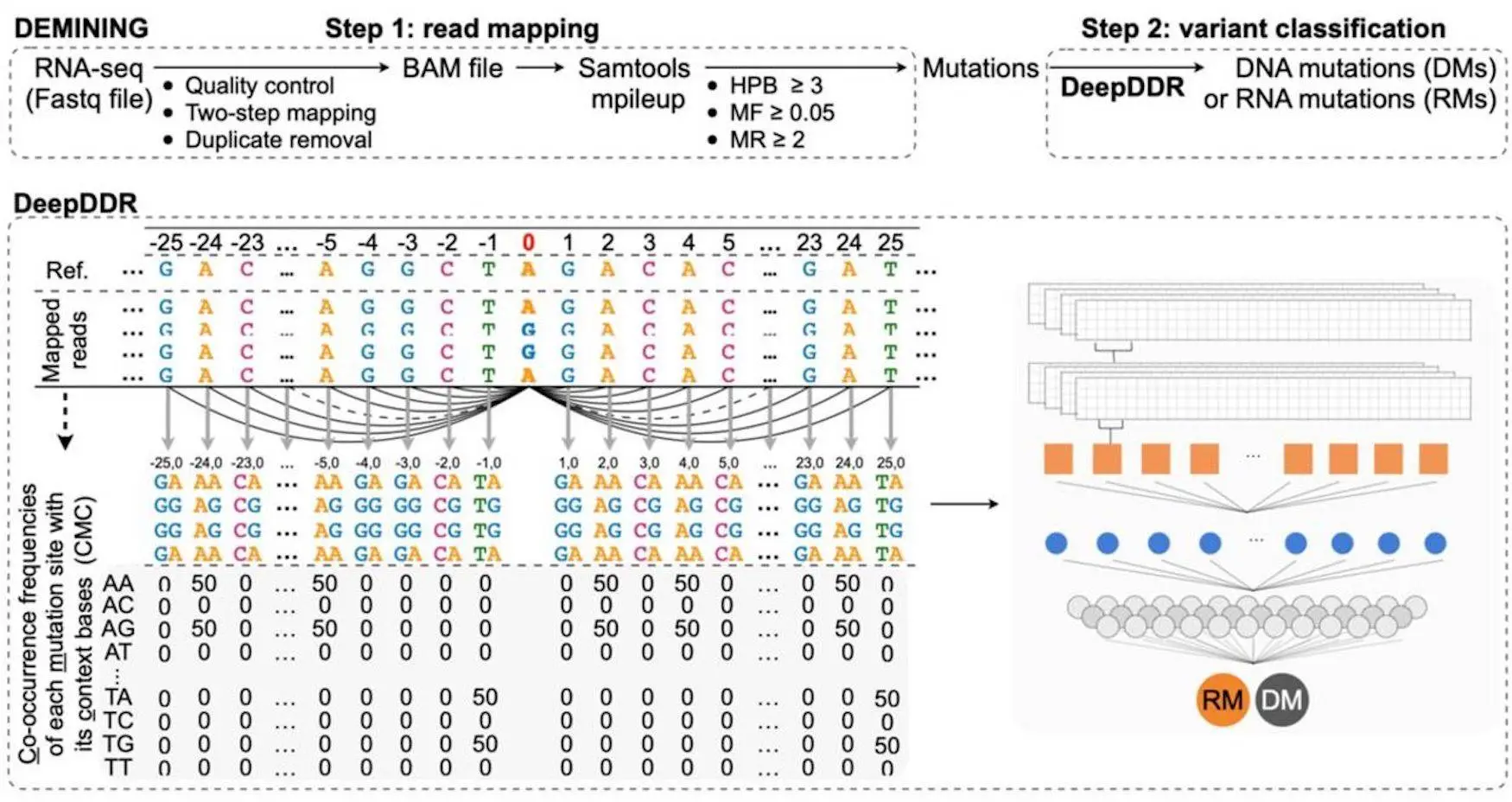
These changes in DNA can be frequently recognized using whole genome sequencing or from RNA-seq data; however, RNA mutations, such as Adenosine-to-Inosine (A-to-I) RNA changes, are difficult to differentiate because of the existence of expressed DNA Mutations. DEMINING, a stepwise bioinformatic technique, was developed to overcome this. DEMINING combines DeepDDR, a sophisticated learning model that distinguishes transmitted DMs from RMs in RNA-seq data. Even working with intense myeloid leukemia (AML) patients, DEMINING uncovered various ignored variations and changes in AML-related gene regions, revealing insight into potential diagnostic and therapeutic targets. This tool provides a new way of recognizing infection-related changes from RNA-seq datasets.
Application of DEMINING to detect AML-associated DMs/RMs from patient samples
The DeepDDR-embedded DEMINING framework is used to analyze publically accessible RNA-seq datasets, mainly focusing on samples from Acute Myeloid Leukaemia (AML) patients. From 19 AML patient datasets, a total of 195,256 Disease-Associated Mutations (DMs) and 137,682 Recurrent Mutations (RMs) were found, showing the diverse character of AML.
To further refine the selection, 57,150 AML-specific DMs were extracted by removing mutations seen in normal control samples or with high minor allele frequencies, highlighting their AML relevance. Notably, 70.91% of these AML-specific DMs coincided with annotated SNPs, although only 13.32% had previously been linked to illnesses in the ClinVar and COSMIC databases. This revealed that several unrecognized DMs might be involved in AML pathogenesis, requiring additional research.
Among the 57,150 AML-specific mutations discovered, 8,220 were recoding ones, possibly generating amino acid alterations in 4,464 genes. Significantly, in the COSMIC Cancer Gene Consensus (CGC), 50 of these genes were linked to AML, with functional involvement in AML formation and progression. These genes shared enhanced biological pathways linked to changes in chromatin and histone modification, which have been found to be misregulated in cancer, according to Gene Ontology analysis.
Association of DEMINING-identified DMs with abnormal gene expression
After conducting research on the effects of recording disease-associated mutations on the host genes and their expression in the setting of Acute Myeloid Leukaemia, the study found an increase of over 15 recording mutations in certain genes. The genes involved were ankyrin repeat domain (ANKRD) gene family members like ANKRD36C, ANKRD36, and ANKRD36B. Surprisingly, these were mostly found in sequences encoding intrinsically disordered regions (IDRs), which have been already linked with disease-causing mutations.
The recoding DMs were linked to an increase in ANKRD gene expression in AML samples, particularly those with DEMINING-identified recoding DMs. This revealed that DEMINING-identified DMs might be functional as cis-expression quantitative trait loci (cis180 eQTLs), while the possibility of indirect effects of DMs on their host gene expression could not be excluded.
DEMINING was extended to non-primate samples via transfer learning
The use of this framework was then applied to samples from non-primate species like mice to address the problem of separating disease-associated mutations and recurrent mutations. These species significantly have different A-to-I RNA editing characteristics than humans. Due to this, the first attempt to use a deep DDR machine learning model based on human data sets produced poor results with a low RM recall rate. Transfer learning was used to overcome this, with the pre-trained DeepDDR model with human data serving as a starting point and then fine-tuning the model using mouse datasets. This DeepDDR-transfer model showed considerable gains in RM prediction accuracy and recall rates.
This strategy was even further verified in a nematode dataset, where it also showed significant improvement in prediction. However, when the fine-tuned DeepDDR-transfer model was used for human data, it resulted in a decline in RM prediction precision and misclassification of certain DMs as RMs. This emphasizes the significance of employing the correct model for the relevant primate or non-primate sample analysis.
Workflow of the Model
This framework has two major phases for directly categorizing Disease-associated mutations and Recurrent mutations from RNA seq data. The first phase identifies high-confidence mutations by aligning and gathering RNA-seq data sets that meet criteria such as base quality, mutation reads, HPB, and mutation frequency. After which, the second step is to feed these high-confidence mutations into DeepDDR. By analyzing the co-occurrence frequency of each mutation, a likelihood score is generated. Mutations with very low probability are classed as DMs, whereas those with high prediction probabilities are classified as RMs.
To check and evaluate the models’ performance, an independent validation set was used, which included 5,348 RMs and 17,230 DMs. For RM prediction, EditPredict and RED-ML were employed, where EditPredict obtained flanking sequences surrounding mutations, and RED-ML was specifically designed for human A-to-I prediction, retrieving features from human genomes.
Gene expression analysis was also done using StringTie and gene ontology analysis to investigate mutation functionality. Stringent criteria were used to accurately differentiate real DMs and RMs in adenosine deaminase knockout and wild-type samples of mass spectrometric data from AML patients.
The best size of the transfer training set that maximizes recall ratio was determined via a systematic selection method. As the final transfer training set, a subset of 500 DMs and 500 RMs was chosen. Based on this subset, the DeepDDR-transfer model was built and fine-tuned using a transfer learning technique. On the transfer test set, the model’s performance was tested.
Evaluation of the Model
DEMINING is a computational model that is designed to differentiate DM from RMs in an RNA-seq data set. It combines high-confidence mutations, obtained using RNA-seq read mapping and filtering, with the DeepDDR model, which is trained on reliable DMs and RMs. After doing the analysis of the 403 RNA-seq datasets, 98% of the discovered RMs were A-to-G and T-to-C mismatches, largely occurring in Alu regions. It shows high-confidence A-to-I RMs. The model successfully integrated these A-to-I RMs from several datasets and made a collection of 122,872 unique RMs and 122,872 unique DMs. The DeepDDR model, when integrated with DEMINING, gives an impressive performance in distinguishing between DMs and RMs.
Conclusion
The demining framework overcomes a major problem in mutation profiling, allowing us to pinpoint DNA and RNA mutations directly in RNA-seq data. It performs better than existing methods like EditPredict and RED-ML and excels in predicting RMs. Its reliability has been proven by its consistent performance across different data sets, making it a valuable tool for research on mutations that are associated with diseases. It is adaptable to a variety of species, which broadens its utility.
This provides valuable information on how these genetic mutations could be linked with the disease. DEMINING’s capacity to directly identify disease-associated mutations offers significant potential for enhancing the understanding of genetic diseases and their diagnosis and treatment.
Article source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Prachi is an enthusiastic M.Tech Biotechnology student with a strong passion for merging technology and biology. This journey has propelled her into the captivating realm of Bioinformatics. She aspires to integrate her engineering prowess with a profound interest in biotechnology, aiming to connect academic and real-world knowledge in the field of Bioinformatics.











{kind=link}