
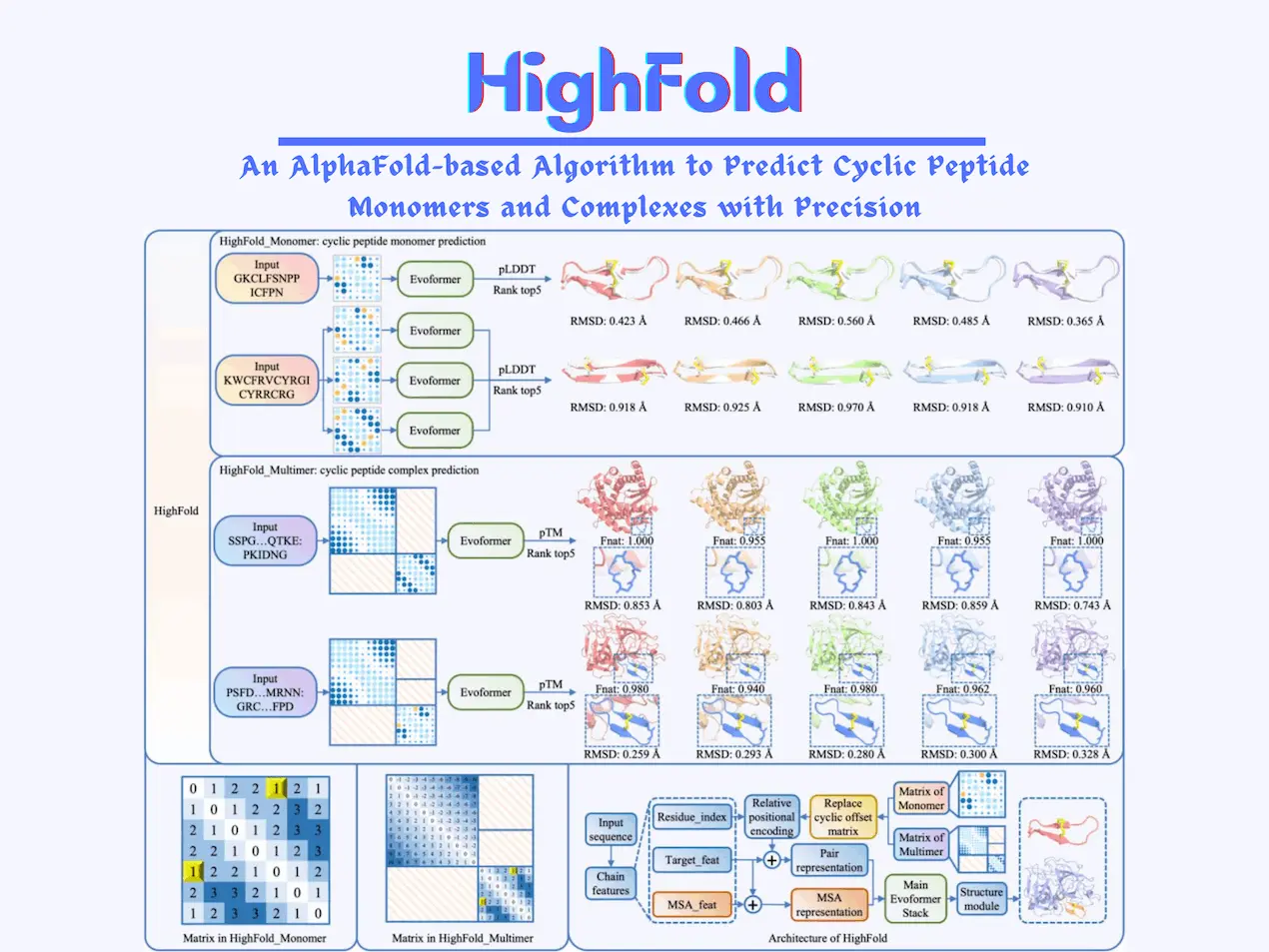
Cyclic peptides represent an emerging class of drugs that combine the advantageous attributes of small molecules with those of antibodies or protein-based therapeutics. Scientists from the College of Pharmaceutical Sciences at the Zhejiang University of Technology and Shanghai Highslab Therapeutics have introduced HighFold, an improved method for predicting cyclic peptide monomers and complex structures. It is based on AlphaFold, a deep learning algorithm used for peptide prediction. It’s hard for deep learning problems to make accurate predictions because they don’t take into account important structural aspects of peptides, especially cyclic peptides, and they only have a small amount of data to work with. HighFold overcomes all of these problems with the incorporation of a novel matrix by the developers.
Importance of cyclic peptides
Cyclic peptides have become a subject of focus in pharmacological studies as they offer several therapeutic uses, owing to the diversity of their biological activities. They can be extensively utilized in drug discovery and development, and their cyclic structures preserve some important features, such as the high binding affinity of the target protein towards the ligand, as well as properties related to the specificity and selectivity of the proteins. These compounds are very adaptive as well. However, they pose challenges in predicting the structural layout of their monomers and complexes, which is vital to enabling further research.
Experimental methodologies that have been traditionally used throughout the years have yielded accurate results but have been very time-consuming and have required a lot of effort and labor. This led to a decrease in efficiency and an increase in cost. Therefore, many computational approaches have been sought to solve these problems.
The first computational approach: Computer Aided Drug Design (CADD)
CADD methodologies have been widely used for drug discovery for years now. The basic principle involved in these methods is molecular docking, wherein proteins (referred to as ‘target proteins’) are predicted based on their interactions with small molecules called ligands. They use a combination of energy functions based on physics and machine learning algorithms like Monte Carlo methods for sampling. However, they require an unreasonable amount of time to determine the structures that have the least energy. The trade-off between the accuracy and efficiency of the resources used is not balanced. Also, peptides that contain more than 30 amino acid residues (known as macrocyclic peptides) are not taken into consideration by most CADD methods. All of these drawbacks lead to unsatisfactory predictions of peptides and proteins.
Some examples of docking tools used in CADD are Rosetta and AutoDock CrankPep (ADCP). Rosetta only analyzes peptides that contain between 5 and 20 residues and runs the data multiple times, between 250 and 50,000 times, to get the desired results. ADCP only analyzes up to 20 residues and is dependent on experimental data related to the structures of target proteins for docking the cyclic peptides that its algorithm generates. It cannot perform docking if insufficient data for the target protein is provided.
Deep-Learning Methods as an alternative solution
Deep learning models have a greater rate of accurately predicting cyclic peptides in a shorter amount of time than CADD methods. AlphaFold and RoseTTAFold are popular examples of such models. Cyclic peptide monomers can be predicted using AfCycDesign, another DL model. However, it ignores the possibility of disulfide bridges being present in the structure, which in turn reduces the accuracy of the cyclic peptide folds predicted by it.
Disulfide bridges, depicted as S-S bonds in many visualizations of peptides and proteins, are important factors to consider when predicting peptides. Conformations of peptides are stabilized through these bonds, and proteases’ increased resistance to degradation is conferred to proteins through these bridges.
HighFold comes into play
HighFold is an algorithm based on AlphaFold that accounts for information related to the disulfide bridges present in cyclic peptide structures and analyzes the circular structure as a whole. It can also predict structures for different lengths of peptides, in contrast to the limitations imposed by Rosetta and ADCP.
It uses matrices as one of the main components of its algorithm. N x N matrices are used for predicting cyclic peptides, where N represents the length of the peptide. The position of each residue is indicated in the matrix; this circularizes the peptide structure and helps in formulating links to disulfide bridges. All possible pairs for disulfide bridges are listed as well.
The matrix containing the collection of bridges is transferred to AlphaFold and AlphaFold_Multimer. The ranking of the structures predicted by the algorithms of the DL models is performed using a scoring method called pLDDT. The top 5 results given by it are ranked and subsequently analyzed.
HighFold consists of two types of models: HighFold_Monomer and HighFold_Multimer. HighFold_Monomer depends on AlphaFold and is used for predicting the structures of monomers. HighFold_Multimer depends on AlphaFold_Multimer and is used for predicting complex structures of cyclic folds. This part gets the additional target proteins as well.
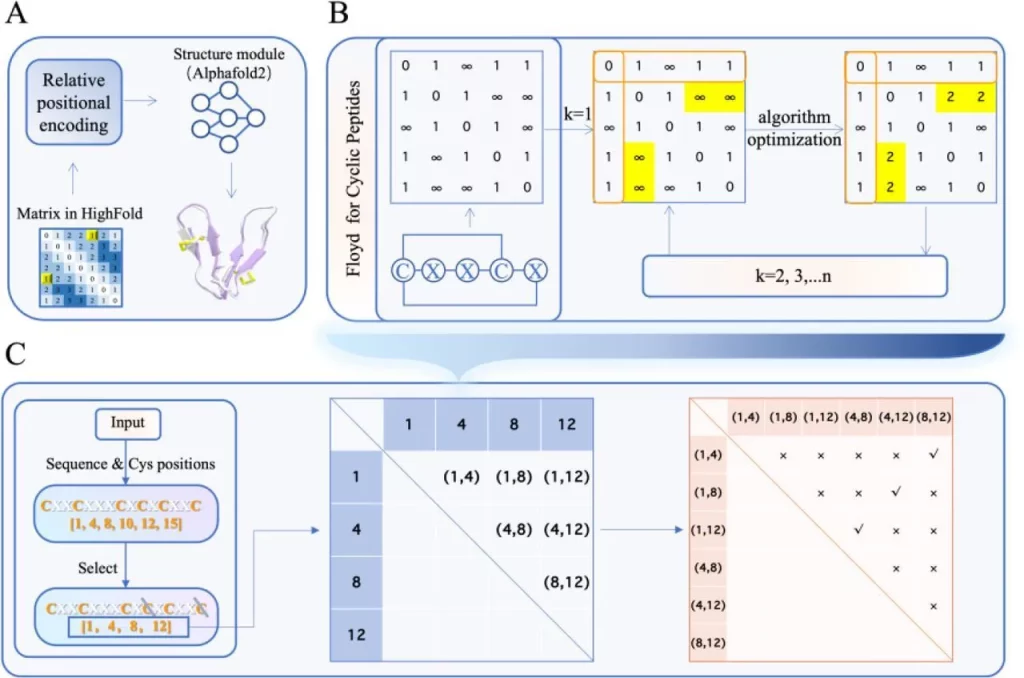
Image Source: https://doi.org/10.1101/2023.08.27.554979
When predicting the structures of peptides and proteins, understanding the nature of interactions between target proteins and ligands that bind to the target protein plays an important role. For differentiating the features of the ligands and target proteins involved, they are separated into two different matrices created for each of them, respectively; the first one is the novel submatrix created by the developers themselves, specifically for the ligand. The second one is the default submatrix used for the target protein.
The dataset used here was retrieved from AfCycDesign. Values of the RMSD (Root Mean Square Deviation) of atomic positions were used to perform several comparisons between the structural predictions given by the HighFold and CADD methods.
Conclusion
HighFold is an automated deep learning approach for cyclic peptide prediction based on the existing algorithms of AlphaFold. It is more refined and accurate as a peptide prediction tool that uses a novel matrix as input to predict complex folds present in cyclic peptides. It is one of the first attempts in bioinformatics to consider disulfide bridges as an important factor in peptide prediction, making it stand out from other methods of prediction. It can predict cyclic structures from head to toe and has the potential to provide insight into the development of different classes of peptides as well. It can accelerate peptide discovery and be of immense importance in the field of drug discovery and development for pharmacological and medical applications.
Article Source: Reference Article
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.





{kind=link}