
Protein structures depend on nucleic acids and tiny ligands to establish binding sites. Accurate identification of these locations is necessary due to the exponential increase of protein sequence databases. However, the current approaches do not thoroughly address the geometry of protein structures and limit genome-scale applications by relying on costly multiple sequence alignments or experimental protein structures. In this publication, researchers from Sun Yat-sen University, China, present GPSite, a multi-task network for simultaneously predicting binding residues of DNA, RNA, peptide, protein, ATP, HEM, and metal ions on proteins. The low-cost binding residue analysis technique GPSite has been utilized to find previously unknown connections between binding sites and biological functions, genetic variations, and molecular activities. On many benchmark datasets, it outperforms the most advanced sequence-based and structure-based methods, even in cases when the structures are not well-predicted. This quick annotation at the genome-scale makes further investigation possible.
Understanding the Landscape of Protein Interaction and Modeling
Proteins interact with other molecules, such as tiny ligands, peptides, nucleic acids, and proteins, to perform essential biological functions. Predicting protein function, comprehending disease mechanisms, and developing new drugs all depend on an understanding of these binding interfaces. Although natural complex structures can be found using X-ray crystallography and mass spectrometry, these methods are expensive, time-consuming, and inappropriate for proteins with unidentified binding partners. Effective computational techniques to identify putative binding sites are essential as sequence databases, including proteins, continue to expand.
Comparative modeling is a conventional method for predicting binding interfaces; it transfers known binding residues from similar templates to the query proteins using alignment algorithms. This tactic is limited, though, if there isn’t a high-quality template. Techniques for machine learning and deep learning have surfaced and categorized into sequence-based and structure-based methodologies. Sequence-based techniques use machine learning classifiers to learn local binding properties from sequence contexts or deep learning models to capture global dependencies. However, the lack of tertiary structural information limits the accuracy of these predictors.
In experimental structure-based methods, binding-relevant spatial patterns are learned by using graph neural networks (GNNs) or geodesic convolutional to encode protein structures as graphs made up of surface point clouds of atoms or residues. However, because of the structure’s undiscovered geometry, most approaches’ expressive potential is still limited. Both sequence- and structure-based predictors confront issues in high-throughput techniques at the genome scale due to their need for multiple sequence alignments (MSA).
Diving into GPSite
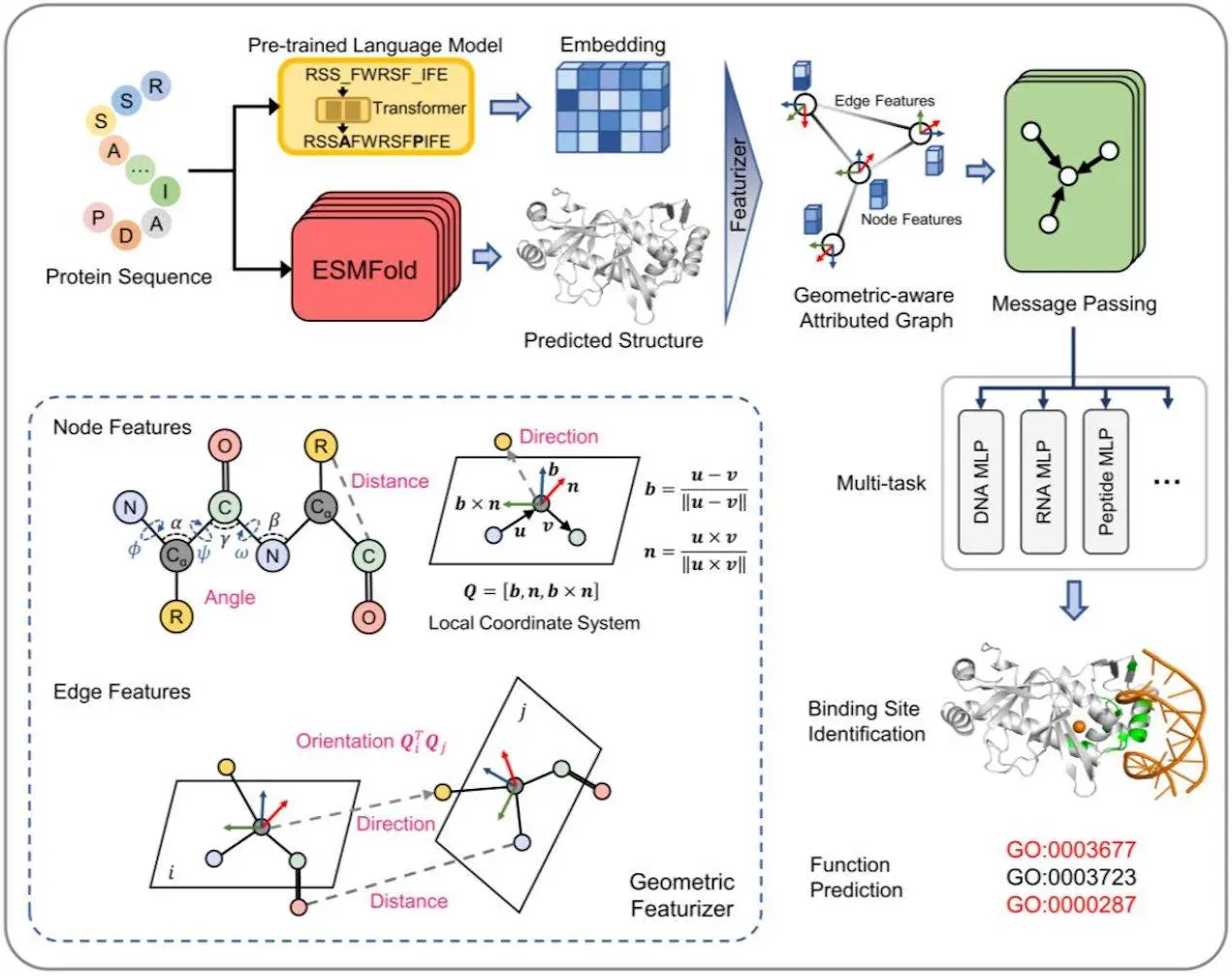
In a multitask framework, GPSite (Geometry-aware Protein binding Site predictor) is a fast, accurate, and versatile network that predicts binding residues of ten different types of biologically relevant molecules simultaneously. These types of molecules include DNA, RNA, peptides, proteins, ATP, HEM, and metal ions (Zn2+, Ca2+, Mg2+, and Mn2+). Because it was trained using informative sequence embeddings and predicted structures produced by protein language models that have already been taught, GPSite is independent of native structures or MSA. A complete geometric featurizer combined with an edge-enhanced graph neural network is designed to extract the residual and relational geometric contexts in an end-to-end manner, hence improving the ability to capture the high-level bio-physicochemical features in the predicted structures.
Tests show that on many benchmark datasets, GPSite significantly outperforms state-of-the-art sequence-based and structure-based methods, even when the predicted structures are of poorer quality. With its fast processing speed, GPSite can handle genome-scale sequence databases, such as the complete Swiss-Prot, and quickly annotate over 568,000 sequences’ binding residues. Subsequent examinations suggest that these annotations may facilitate the identification of connections between binding locations and biological mechanisms, genetic variations, and molecular activities.
Key Findings
1. Identification of Novel Binding Sites: Protein binding residues may be precisely identified from protein sequences using GPSite, a predictor of protein binding sites. To produce informative sequence embedding and anticipated structure, it makes use of the folding model ESMFold and the pre-trained language model ProtTrans. The centroid of the heavy sidechain atoms and the coordinates of the N, Cα, C, and O atoms are included in the anticipated structure. For every residue, the DSSP determines the secondary structure and relative solvent accessibility. Residues are connected as nodes, and neighboring nodes are connected as edges in a protein radius graph. An end-to-end geometric feature extractor is developed to create a local coordinate system for every residue and extract geometric characteristics that capture sidechain and backbone atom configurations. Intra-residue distances between two atoms, sidechain centroid or other inner atoms’ relative orientations to Cα, bond and torsion angles, and spatial orientations between neighboring nodes are examples of geometric node properties.
GPSite is a GNN that takes into account multi-scale interactions at the node, edge, and global context levels in order to learn residue representations. On a geometrically aware attributed graph, it makes use of message passing, edge update, and global node update techniques. The message-carrying layer updates the central node by aggregating data from the nearby neighborhood using multi-head attention in the transformer boosted by edge characteristics. Gated attention is used to update the node states using global context information, and the features of an edge are changed using the nodes that connect it. GPSite employs a multi-task learning technique, rotationally and translationally invariant, and shares the GNN among various ligands to capture common binding-relevant properties. Through concurrent prediction, this framework shortens the time required for the inference of numerous ligands. Using ProtTrans and ESMFold, allowing genome-wide binding site prediction, taking advantage of a thorough geometric featurizer, and using message propagation on residue graphs are some of the ways that GPSite differs from earlier methods. Additionally, it makes use of multi-task learning to enhance the modeling of latent interactions between several binding partners.
2. Unprecedented Annotation Accuracy: The Swiss-Prot database was subjected to annotation and analysis of possible binding interfaces using GPSite, a computational tool. It took about 8.5 days for researchers to use ESMFold to predict the structures of 568,326 sequences. About five hours were spent on feature extraction and GPSite inference methods. All Swiss-Prot binding site annotations and ESMFold-predicted structures are included in the user-friendly database GPSiteDB. To distinguish between proteins that bind and those that do not, the researchers employed residue-level binding site annotations for a variety of ligands. For every ligand, a protein-level binding score was produced by averaging the top k predicted values over all residues. Proteins having related ligand-binding molecular functionalities in Swiss-Prot have substantially better GPSite binding scores than proteins without such annotations.
ROC curves (receiver operating characteristic curve) were used to validate the accuracy of GPSite protein-level binding scores. All ligands except protein had adequate AUC values. To increase the number of binding function annotations in Swiss-Prot, the top 20,000 proteins with the highest GPSite binding scores were compiled. It was discovered that DNA repair, DNA-templated transcription, DNA recombination, and replication are among the biological processes in which GPSite-predicted binding proteins are implicated.
Cell researchers can detect genetic variations and evaluate their possible disruptions in protein-ligand interactions and pathogenicity thanks to the predicted structures found in GPSiteDB. The research demonstrates that 20.67% of pathogenic variant sites on the surfaces of anticipated structures fall in GPSite-predicted interfaces, greater than benign variants (11.97%) or the random baseline (16.35%). Variants in predicted binding sites have a higher pathogenic probability than variants in non-binding sites. Remarkably, mutations in the anticipated binding sites of metal ions and ATP have higher pathogenic probabilities than variants in other ligands. This may be because the binding interfaces’ tiny pockets are created by a small number of residues.
3. Insights into Functional Significance: Protein Data Bank (PDB) binding site benchmark datasets were used to train and assess GPSite, a protein-based prediction model. For ten different ligand types, GPSite obtained average AUC values of 0.918 and 0.921, as well as AUPR values of 0.603 and 0.594 in independent and cross-validation tests. GPSite surpassed all other methods in AUPR for more than 16.5%, 13.2%, 55.4%, 1.7%, 27.7%, 10.8%, 7.0%, 14.8%, 17.1%, and 13.4% in the DNA, RNA, peptide, protein, ATP, HEM, Zn2+, Ca2+, Mg2+, and Mn2+ binding site test sets, in comparison to eight sequence-based predictors and fifteen structure-based predictors. Additionally, GPSite can use native structures as prediction input, increasing its average AUPR by 7.8%. However, the efficacy of these approaches diminishes dramatically when employing predicted structures for testing, as they were trained with high-quality native structures. This further demonstrates GPSite’s advantage in real-world scenarios where experimental constructions are not accessible.
A solid tool that does a better job of predicting structures than GraphBind is GPSite, a model based on ESMFold. For a variety of datasets, ESMFold achieves median TM-scores of 0.89, 0.76, 0.93, 0.93, 0.94, 0.95, and 0.96. On low-quality predicted structures, GPSite performs similarly to GraphBind, with a TM-score ≤ 0.7 indicating inferiority. But even in low-quality scenarios, GPSite regularly performs better on predicted structures than GraphBind. A case in point is the human glucocorticoid receptor (GR) structure, which shows that GPSite can distinguish between different interfaces even in cases where the structure is not exactly expected. With AUPR values of 0.949 and 0.924, respectively, GPSite can accurately identify the binding sites of DNA and peptides and can properly anticipate all Zn2+ binding sites. In general, GPSite’s performance is greatly influenced by how reliable it is at predicting structures.
Future Direction
The GPSite model, a tool for predicting protein structures, has made significant progress. The ESM Metagenomic Atlas offers 772 million predicted protein structures and pre-computed language model embeddings. Self-supervised learning can be used to train a GPSite model for predicting masked sequence and structure attributes. The model can maximize similarity between learned representations of substructures from identical proteins while minimizing similarity between those from different proteins using a contrastive loss function. Fine-tuning this model on binding site datasets may improve performance. Additional opportunities for improvement include a variational Expectation-Maximization framework to handle the hierarchical graph structure inherent in proteins, allowing for interaction and mutual enhancement between modules. Meta-learning can also be explored for fast adaptation to unseen tasks with limited labels.
With the unmatched speed at which unannotated and annotated sequences are becoming separated, GPSite is a dependable, effective, adaptable, and user-friendly tool for deciphering the vast and ever-changing field of protein-ligand interactions. By utilizing GPSite’s capabilities, scientists can easily discover new biological roles for proteins, learn important lessons about the underlying pathogenic mechanisms of gene mutations, or create innovative medications that target certain binding sites.
Conclusion
GPSite is a new technique that can precisely predict the protein binding sites of a wide range of biological substances, such as metal ions, ATP, DNA, RNA, peptides, and proteins. To give high-level bio-physicochemical features, it makes use of expected structures and language models that have already been trained. GPSite refines residual and relative geometric contexts using an edge-enhanced graph neural network and a complete geometric featurizer. Additionally, it models intrinsic interactions among binding partners using a multi-task framework. The outcomes demonstrate that GPSite performs better than cutting-edge techniques, even with projected structures of lesser quality. Annotating binding sites for approximately 568,000 sequences in Swiss-Prot in less than nine days demonstrates GPSite’s scalability and helps uncover previously undiscovered biology related to protein function and genetic variation.
Article Source: Reference Paper | GPSite: Webserver
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}
[…] Language Models Paired with Geometric Deep Learning Revolutionize Genome-scale Protein Binding Site … […]