
A deep learning-based method for predicting the biophysical and biochemical features of protein-ligand complexes developed by researchers could help discover new drug candidates.
Scientists formulate novel parallel graph neural networks for decoding protein-ligand interactions. This methodology can serve as an interpretable and explainable artificial intelligence (AI) tool for predicted activity, potency, and biophysical properties of lead candidates.
Image Source: Decoding the protein-ligand interactions using parallel graph neural networks.
Protein-ligand interactions (PLIs) are fundamental for biochemical functionality, and their identification is essential for assessing biophysical properties for rational therapeutic design.
Currently, experimental characterization of the properties is the most reliable strategy, nonetheless, it is exceptionally tedious and labor-intensive. Various computational approaches have been developed in this setting, but a vast majority of the current PLI prediction heavily relies upon 2D protein sequence data.
This study presents a novel parallel graph neural network (GNN) to integrate knowledge presentation and reasoning for PLI prediction to perform deep learning directed by expert knowledge and informed by 3D structural data.
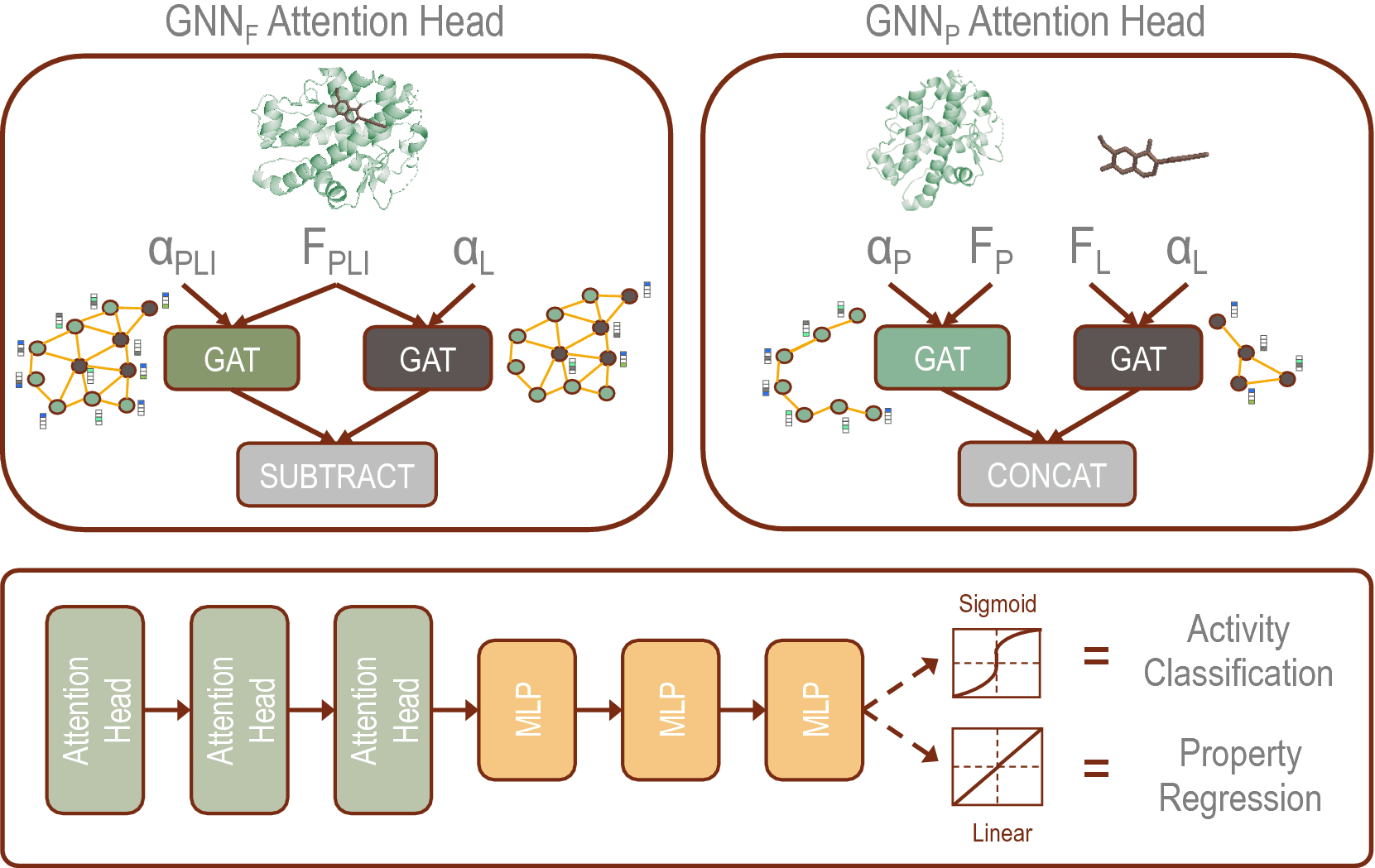
The scientists developed two distinct GNN architectures:
- GNNF is the base implementation that utilizes distinct featurization to enhance domain awareness while
- GNNP is a unique implementation that may predict intermolecular interactions without prior knowledge.
The comprehensive assessment demonstrated the way that GNN can effectively capture the binary interactions among ligand and protein’s 3D structure with 0.979 test accuracy for GNNF and 0.958 for GNNP for predicting the activity of a protein-ligand complex.
These models were additionally adapted for regression tasks to predict experimental binding affinities and pIC50 significant for the compound’s potency and efficacy.
The scientists achieved a Pearson correlation coefficient of 0.66 and 0.65 on experimental affinity and 0.50 and 0.51 on pIC50 with GNNF and GNNP, respectively, outperforming similar 2D sequence-based models.
This methodology can serve as an interpretable and explainable artificial intelligence (AI) tool for predicted activity, potency, and biophysical properties of lead candidates.
To this end, the scientists demonstrated the utility of GNNP on SARS-Cov-2 protein targets by screening a huge compound library and comparing the prediction with the experimentally measured data.
The Utilization of Molecular Docking in Binding Affinity Prediction
Accuracy in protein-ligand interactions (PLI) prediction is a basic advance in therapeutic design and discovery. These interactions impact different molecular-level properties, for example, substrate binding, product release, regio-selectivity, target protein function, and ability to facilitate potential hit identification, which is the initial step in finding novel candidates for drug discovery.
With expansions in computing power, code scalability, and advancement of theoretical methods, physics-based computational tools like molecular dynamics and molecular/quantum mechanics can be utilized for the reliable representation of PLI and prediction of accurate binding free energies.
Nonetheless, these methodologies are computationally expensive and are restricted to several protein-ligand complexes. This limits their routine utilization in high-throughput virtual screening for the discovery of novel hit candidates and lead optimization for a given protein target.
Molecular docking has been utilized to predict binding affinity and estimate interactions with reasonable computational cost; its precision is relatively low as it utilizes heuristic rules to assess the scoring function.
The Utilization of Deep Learning Models for the Prediction of PLI
The utilization of deep learning has revolutionized the healthcare framework in recent years. Significant effort has been laid to develop deep learning models that predict PLI and other biophysical properties that are basic for therapeutic design yet can’t be predicted through physics-based modeling.
The more prominent comprehension of PLI empowered by deep learning can help in the estimation of properties, for example, activity, potency, and binding affinity. Be that as it may, a few technical difficulties limit the utilization of deep learning for modeling protein-ligand complexes and accurate prediction of properties.
The first challenge connects with the restricted availability of protein-ligand 3D information, and the second challenge centers around the appropriate representation of the information (domain knowledge), explicitly concerning the comprehensive 3D geometry representation.
Structure-based methods have the benefit of producing results that can be interpreted but are restricted by the number of available samples. Circular fingerprints, which are created by encoding localized structural and geometric data, have been a cornerstone of cheminformatics for a long time.
The flexibility of fingerprints has made new avenues for molecular computational research, including the increased implementation of graph-based representations to include domain awareness.
Molecular graph representations give way to modeling and stimulating the 3D chemical space while retaining a more extensive scope of structural data. In that unique circumstance, a deep convolutional neural network (CNN) was implemented that operated straightforwardly on 3D molecular graph input, like the AtomNet model similarly carried out.
Other different methodologies, for example, Graph-CNN, utilize unsupervised autoencoders to leverage sequence-based information that is more abundant yet additionally expensive in terms of structural accuracy.
Graph-Based Representations and their Role in Chemical Data Learning
Graph-based representations expand the learning of chemical information to graph neural networks (GNNs). So, a mixture model network (MoNet) that enabled non-Euclidean data, like graphs, to be learned by CNNs, was designed.
That approach has been summed up and improved through the formulation of graph attention (GAT) networks. GAT architectures work on the significance of a given node, prompting improved computational effectiveness and accuracy.
Protein-ligand complex activity prediction is a binary classification problem. Reshaping the issue to focus on affinity creates a regression problem of heightened complexity.
The current deep learning models predict the binding affinity or other biophysical and biochemical properties like IC50, Ki, Kd, and EC50.
Be that as it may, the vast majority of the strategies use sequence-based data for proteins and SMILES representations for interacting ligands. For instance, DeepDTA and DeepAffinity use SMILES strings of the ligands and amino-acid sequences of the target proteins to predict the affinity.
MONN is a multi-objective sequence-based neural network model that initially predicts the non-covalent interaction between the ligand and the residues of the interacting target and, afterward, the binding affinities regarding IC50, Ki, and Kd.
Such strategies are accessible because of the abundant availability of sequence-based data, however, they don’t capture 3D structural data in the PLI and predict regression properties.
Binding is best perceived when the 3D pocket of the target is known, and in situ, the protein-ligand complex is formed because of changes in the conformation of the 3D structure of the protein and ligand post-translation.
Formulation of New GNNs and their Role
In this study, the scientists formulated two GNNs because of the GAT architecture by integrating domain-specific featurization of the protein and ligand atoms (GNNF) and by implementing parallel GAT layers to such an extent that GNNP uniquely learns the interaction with limited prior knowledge.
The addition of various features on the protein and ligand atoms enables the models to be more physics informed. The execution of GAT layers joined with the featurization allows the model to become familiar with the training data’s representation and the chemical space.
The scientists further utilized these models to predict the protein-ligand complex’s experimental binding affinity and pIC50. This permitted the use of 3D structures of the target protein, ligands, and the interaction between them, which is vital for activity and affinity prediction.
The Endpoint
In this study, the scientists devised graph-based deep learning models, GNNP and GNNF, by incorporating knowledge representation, 3D structural information, and reasoning for PLI prediction through classification and regression properties of protein-ligand complexes.
The parallelization of the GNNP model gives a premise for novel implementation of structural analysis that requires no docking input except for different protein and ligand 3D structures.
The fundamental strategy of GNNP is to learn embedding vectors of the ligand graph and protein graph independently and join the two embedding vectors for prediction. The featurization of GNNF gave a baseline to their implementation of domain aware capabilities enhanced through feature engineering to identify critical nodes and differentiate the contribution of each interaction to the affinity.
In GNNF, the embedding vectors are learned simultaneously for the protein and ligand complex as an early embedding strategy.
Through the study, we understand that the model is unique and new in that it uses the protein and ligand’s 3D structures to estimate affinity and pIC50, which is critical for determining a candidate’s potency and selectivity to inhibit a particular protein target, which is vital to give a quantitative measure of the potency and selectivity of a candidate to inhibit a specific protein target.
To speed up in silico hit identification and lead optimization in the beginning phase of drug design, the GNNP model can be used to screen a huge ligand library for biophysical properties or activities against a specific protein target or a set of targets for a specific disease.
Article Source: Knutson, C., Bontha, M., Bilbrey, J.A. et al. Decoding the protein–ligand interactions using parallel graph neural networks. Sci Rep 12, 7624 (2022). https://doi.org/10.1038/s41598-022-10418-2
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}