
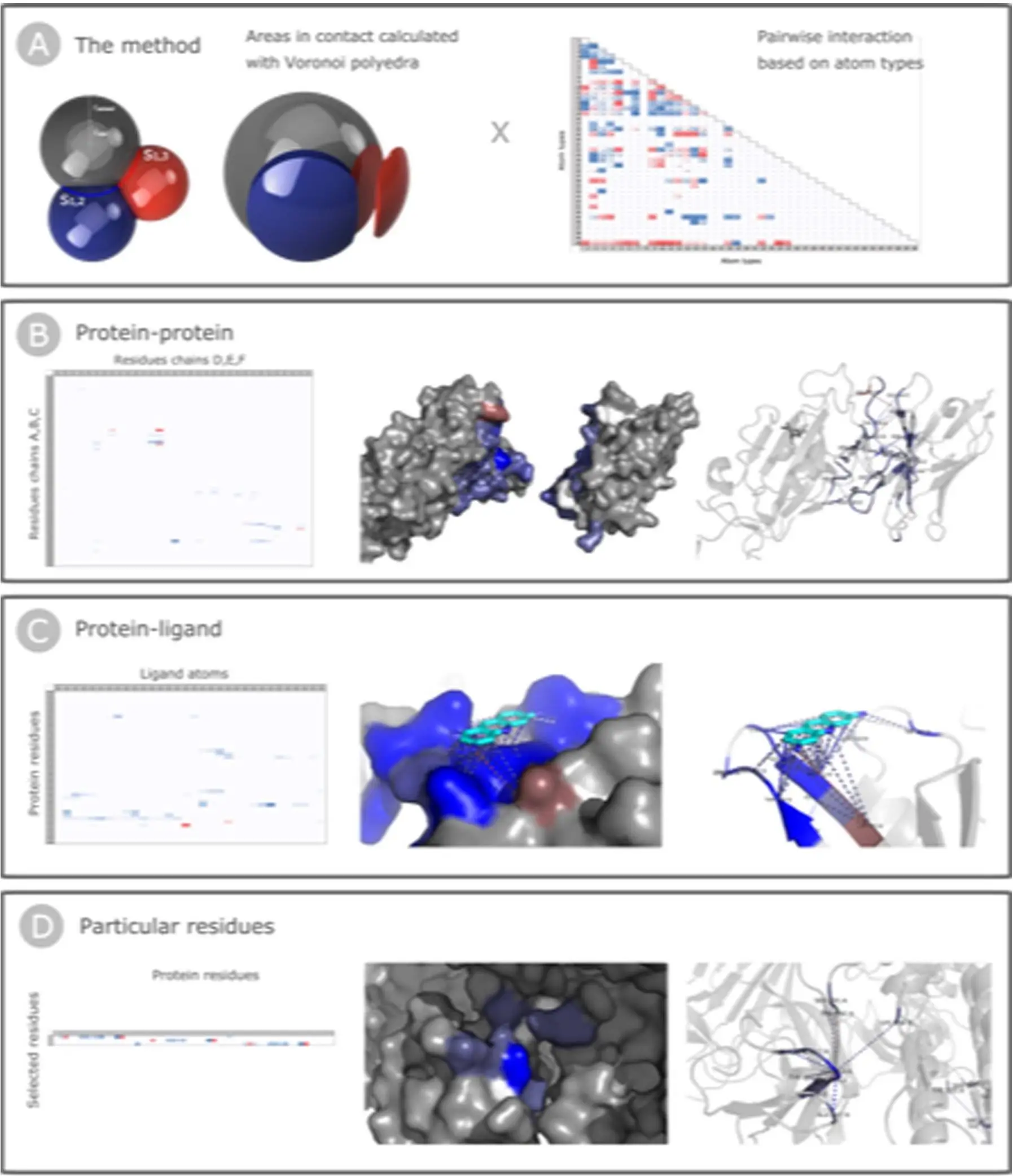
Computational methods have revolutionized our understanding of molecular interactions and their pivotal role in the stability of biomolecular structures and complexes. These insights are crucial for modulating, engineering, and comprehending biological processes. Scientists from the Université de Montréal, Canada, introduce Surfaces, a groundbreaking software that offers a seamless, fast, and highly customizable approach to quantify and visualize molecular interactions. This innovative tool is based on the calculation of surface areas in contact, demonstrating equivalent or superior correlations with experimental data compared to computationally intensive methods relying on molecular dynamics.
Protein engineering is a cornerstone of biotechnology, enabling the creation of proteins with enhanced therapeutic and industrial applications. Despite significant progress in recent years, the immense search space of potential sequence configurations and corresponding structures remains a formidable challenge. Random exploration of this space is impractical, necessitating a rational design approach. Techniques that unveil the individual contributions of atoms or residues to protein interactions are invaluable in devising effective protein engineering strategies. However, many of these techniques, predominantly based on ab initio approaches, are computationally demanding, limiting their application in large-scale studies.
With the decreasing cost of computational power, it has become feasible to simulate biological systems with greater fidelity to the underlying physical processes governing molecular interactions. Molecular dynamics (MD) simulations stand at the forefront of these endeavors. However, MD methods are computationally expensive and often pose implementation challenges, rendering them impractical for high-throughput applications or widespread adoption. Notable methods in this domain include FoldX, Rosetta, free-energy perturbation (FEP), and molecular mechanics methods (MM/PBSA and MM/GBSA), each aimed at predicting binding energies for different mutations or biomolecular complexes.
To address this challenge, simplified techniques utilizing atomic surface areas in contact have emerged as a viable alternative for estimating binding energy. While these methods, such as LPC (Ligand-Protein Contacts), CSU (Contacts of Structural Units), and STC (Structure-based Thermodynamic Calculations), have shown promise, limitations in server availability, atom type definitions, and energetic matrices persist. Nonetheless, they serve as the foundation upon which subsequent methods have been built, highlighting the potential for further refinement and customization.
Surfaces: A Game-Changing Tool
In response to this need, the researchers introduced Surfaces, a swift method leveraging atomic surface areas in contact, user-defined atom-type definitions, and pairwise pseudo-energetic matrices. This approach provides a proxy for identifying favorable and unfavorable interactions within and between proteins, as well as between proteins and other biomolecules.
Surfaces employ two measures to quantify atomic interactions. The first measure calculates the area in contact between atoms using a Voronoi procedure. The second measure involves a pairwise pseudo-energetic matrix, assigning an interaction pseudo-energy based on atom types. The complementarity function (CF) combines these measures, yielding valuable insights into the contributions of individual atoms or residues.
Applications of Surfaces
Surfaces can analyze various types of interactions, including protein-protein, protein-ligand, and residue interactions. The output provides quantitative data in CSV format and visual representations in PyMOL session files. Surfaces require a pre-processing step to clean the input PDB structure of any non-defined atoms, offering users the flexibility to customize the analysis.
Validation of Surfaces
Surfaces have been rigorously validated against established methods, demonstrating comparable or superior performance. For instance, using the AB-Bind dataset, Surfaces showcased correlations similar to leading methods like Rosetta and FoldX. Additionally, in a highly curated dataset of SARS-CoV-2 Spike RBD/ACE2 binding ΔΔG values, Surfaces exhibited a Pearson’s correlation of 0.556, rivaling the performance of FEP + 100 ns MD.
Conclusion
Surfaces represent a transformative leap in the analysis and visualization of biomolecular interactions. Its scriptable and customizable nature empowers researchers to explore large structural datasets, opening new frontiers in virtual screening and protein engineering. By offering an accessible, low-resource-demanding solution, Surfaces democratizes the study of molecular interactions, enabling a broader scientific community to unravel the complexities of biological processes. With its unparalleled performance and ease of use, Surfaces stands poised to drive unprecedented advances in protein engineering and computational biology.
Article source: Reference Paper
Learn More:



Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}