
Artificial intelligence (AI) has significantly changed the scientific research landscape in recent years, yet structure-based drug development has not been significantly affected. However, a promising development comes from German scientists in the form of the MISATO dataset, a curated collection of protein-ligand complexes, molecular dynamics traces, and electronic properties. This article explores the significance of MISATO and how it can pave the way for powerful AI-based drug discovery tools.
AI predictions, exemplified by AlphaFold 2, have transformed various scientific fields. AlphaFold’s accurate protein structure predictions have brought it close to state-of-the-art experimental data. However, despite these achievements, the field of structure-based drug discovery still faces challenges. Developing new drugs using structural methods is complex, and current approaches suffer from low precision and computational limitations.
The Need for Curated Datasets
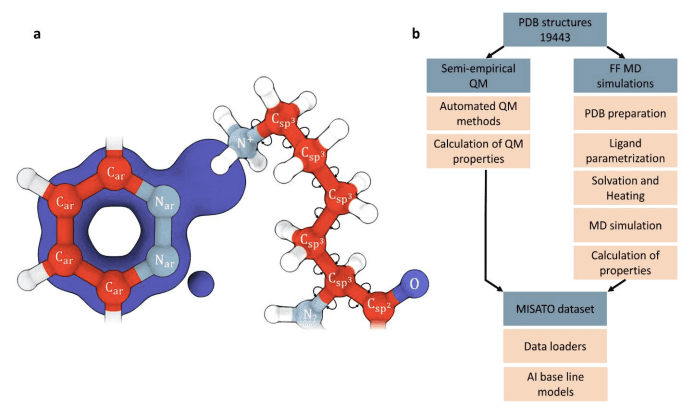
To train AI models for structure-based drug discovery, highly curated and precise biomolecule-ligand interaction datasets are crucial. MISATO fills this gap by providing nearly 20,000 experimental structures of protein-ligand complexes, along with molecular dynamics traces and electronic properties. The dataset is carefully refined using semi-empirical quantum mechanics and explicit water simulations, ensuring accuracy and reliability.
MISATO is designed to be conveniently accessible to the scientific community. With simple Python data-loaders, researchers can access the dataset and incorporate it into their AI models. Additionally, AI baseline models for dynamical and electronic properties are provided, simplifying the adoption of MISATO for drug discovery research.
Overcoming Limitations of Structural Datasets
Structural databases have limitations due to factors such as limited spatial resolution and processing software inaccuracies. MISATO addresses these issues by refining and curating protein-ligand structures. By correcting atom assignments, resolving hydrogen atom issues, and regularizing ligand geometries, MISATO ensures the correctness and consistency of the dataset.
Quantum Chemical-based Curation of Ligand Space
The refinement of ligand structures in MISATO is achieved using semi-empirical quantum chemical methods. These methods strike a balance between computational efficiency and accuracy, making them suitable for curating a diverse range of ligand structures. The curation process involves identifying and correcting inconsistencies in protonation states, electron counting, and molecular charges.
Evaluation and Properties of the Curated Ligands
MISATO’s curation process resulted in modifications to approximately 20% of the original structures. Standard adjustments included removing hydrogen atoms and addressing incorrect atom assignments. The curated ligands were then subjected to various calculations, yielding molecular and atomic properties such as electron affinities, chemical hardness, polarizabilities, and more. These properties provide valuable insights for AI models in drug discovery.
MISATO’s curated dataset and associated properties lay the foundation for harnessing the power of AI in structure-based drug discovery. By providing a comprehensive and reliable dataset, AI models can learn from the fundamental state variables that describe protein-ligand interactions. This opens up avenues for advanced AI approaches, surpassing the limitations of current methods.
Transforming MISATO into a Community Project
The vision for MISATO extends beyond a curated dataset. The researchers aim to foster collaboration and create a vibrant community project for developing robust AI-based drug discovery tools. By encouraging contributions, expanding the dataset, and continuously improving AI models, MISATO aims to accelerate the discovery of novel drugs and revolutionize the field.
Conclusion
Artificial intelligence can potentially revolutionize structure-based drug discovery, and MISATO serves as a crucial step in unlocking that potential. With its curated dataset, refined ligand structures, and comprehensive properties, MISATO paves the way for the development of powerful AI models. By combining AI’s predictive capabilities with accurate and reliable data, researchers can overcome the limitations of current methods and expedite the discovery of life-saving drugs. The future of drug discovery lies at the intersection of AI and curated datasets like MISATO, where innovation and collaboration hold the key to transforming scientific research and improving human health.
Article Source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:



Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.





{kind=link}