
To understand how living systems operate and develop new medications, it is essential to identify protein functions accurately. Researchers from the USA propose a new model, ATGO, for predicting Gene Ontology (GO) attributes of proteins using a triplet neural network that is enhanced with unsupervised language models trained on protein sequences. The integration of the language model with the triplet neural network allows for improved prediction of GO attributes, a valuable resource for understanding protein function and regulation.
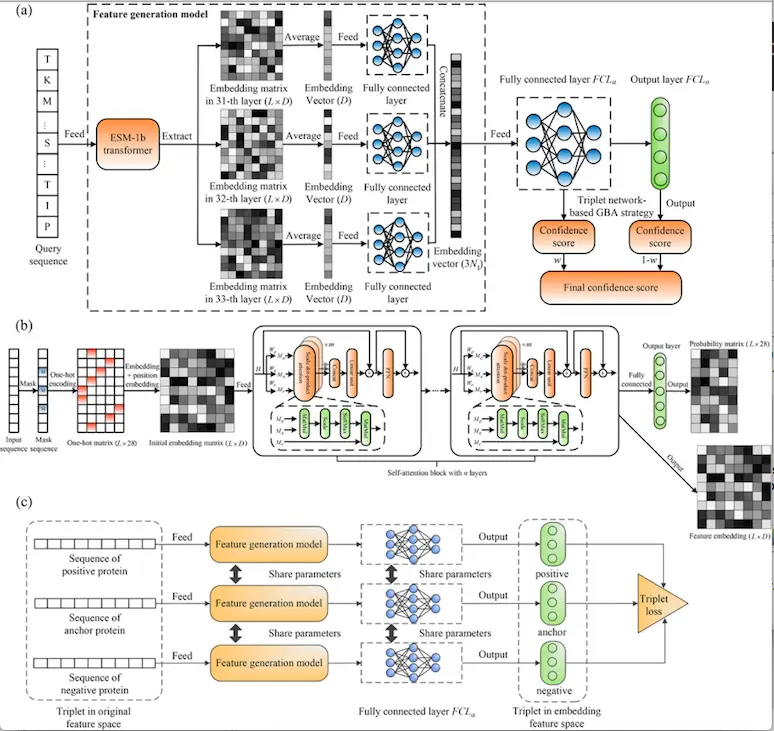
Image Source: https://doi.org/10.1371/journal.pcbi.1010793
The technique was thoroughly evaluated using 3328 targets from the third Critical Assessment of Protein Function Annotation (CAFA) challenge and 1068 non-redundant benchmarking proteins. According to experimental findings, ATGO significantly improved GO prediction accuracy when compared to state-of-the-art methods in all facets of molecular function, biological function, and cellular components.
A thorough analysis of the data revealed that the key benefit of ATGO is the use of pre-fixed transformer language models that can infer discriminative functional motifs from the attribute datasets. The sequence embedding space’s ability to associate functional similarity with feature similarity is improved by the suggested triplet network. It was also discovered that the accuracy of the projected models might be increased by combining the network scores with complementary homology-based conclusions. These findings showed a new method for large-scale protein function annotations from sequence alone using high-accuracy function prediction of deep learning model.
The annotation of all genes and gene products’ biological roles, which have been divided into three criteria: molecular function, biological process, and cellular component, represents a significant issue for computational molecular biology in the post-genome sequencing age. In this study, GO words for proteins were inferred from the main amino acid sequence using a novel open-source deep learning model called ATGO that combined a triplet neural network with previously taught language models of protein sequences. Extensive benchmark testing revealed that, when supported by transformer embeddings of the language model, ATGO greatly outperformed other futuristic techniques in all GO aspect predictions. This study proved the substantial potential of attention transformer language models on protein function annotations, which follows the accelerated development of intra-attention neural network approaches exhibiting impressive outcomes in natural language processing, multi-sensory data processing, and most notably, protein structure prediction.
Proteins, the building blocks of life, have several critical functions in living things, including signal transmission, cellular structure maintenance, and catalyzing metabolic events. Identification of proteins’ biological activities, which have been categorized into three categories by the widely used Gene Ontology (GO)—molecular function (MF), biological process (BP), and cellular component—is essential to understanding life mechanisms (CC). Directly determining the activities of proteins by biochemical assays is common but frequently time-consuming and insufficient.
The three data sets used by existing function prediction techniques are template identification, biological systems, and sequence structure. The discovery of templates that are ideal for functional inference and have a sequence or structure comparable to the query is a common strategy used in conventional approaches for function prediction. For instance, COFACTOR and ProFunc use structural alignment to find templates, whereas GOtcha, GoFDR, and Blast2GO discover sequence templates using BLAST or PSI-BLAST conformations.
In this study, researchers introduced the ATGO deep learning model, which combines the triplet neural network protocol with protein sequence language models to predict protein function with excellent accuracy. Using an unaccounted intra-attention transformer model, ESM-1b transformer, as a pattern extractor to produce feature embeddings initially, this model conjured its foundation on millions of unannotated sequences. The difference between positive and negative samples was then increased using a supervised triplet neural network to train function annotation models from multi-layer transformer feature embeddings. In order to increase model efficacy, a composite version, ATGO+, by fusing ATGO with a sequence homology-based model was also framed. The results showed a considerable improvement in accurate GO word prediction above the existing advanced field for both ATGO and ATGO+ compared to a large range of non-redundant proteins.
Workflow of Protein Function Prediction Model ATGO
(a) The ESM-1b transformer is used to create by fusing feature embeddings a fully connected triplet neural network starting with the input sequence obtained from the last three layers. The confidence scores of GO models are then generated using the fused feature embedding that was input into a triplet network.
(b) The ESM-1b transformer’s construction. The masking, one-hot encoding, and position embedding operations are sequentially carried out for an input sequence to produce the coding matrix, which is then fed into an n-layer self-attention block. By merging m attention heads with a feed-forward network, each layer may produce a feature embedding matrix from a single evolutionary perspective, where each head performs scale dot-product attention.
(c) Creation of a triplet network to evaluate the similarity between features. The input is a triplet variable (anc, pos, neg), where pos (or neg) is a positive (or negative) protein with the same (or different) function as anc, and anc is an anchor (baseline) protein. Each sequence is transmitted into the feature generation model to obtain a feature embedding vector, which is then used as the input of a fully linked layer to produce a new embedding vector. The Euclidean distance of embedding vectors is then used to calculate the feature dissimilarity between two proteins. In order to strengthen the connection between functional similarity and feature similarity in embedding space, a triplet loss is also constructed.
ATGO’s Approach for Protein Function Prediction
The ATGO approach, which uses deep learning to predict protein functions, takes a query amino acid sequence as input and outputs confidence ratings for the predicted GO keywords. Three processes make up ATGO: triplet network-based function prediction, neural network-based feature fusion, and multiple-view feature extraction using a transformer. The first and second processes are together referred to as feature generation models (FGM).
Method I: Recovery of multiple-view features using a transformer.
Method II: Neural network-based feature fusion.
Method III: Triplet network-based function prediction.
Countless developments have contributed to ATGO’s progress. The ESM-1b transformer can successfully extract the discriminative feature embeddings for the input sequence from the many perspectives of evolutionary diversity, which is the first and most crucial step. Second, the multi-view feature fusion lessens the harm brought on by feature extraction information loss. The triplet network-based GBA method, which maximizes the feature distance between positive and negative samples, is crucial to improving training efficiency. Last but not least, using ATGO with supplementary data from sequence homologous inference can boost prediction precision even more.
Conclusion
Considering the transformer’s proven efficacy, it’s vital to remember that the ESM-1b transformer now in use is just one of several language models that have been created based on a single query sequence. Future research should examine various embedding language models for GO prediction, such as ProtTrans and the recently published expanded version ESM-2, both of which outperformed ESM-1b. We will also create a new unsupervised protein language model by using the MSAs produced from DeepMSA through self-attention networks, given that MSA holds considerably supplemented evolutionary documentation labeled with conserved protein sites, which are essential to protein functional annotations. The embedding should further enhance the selectivity and reliability of the GO prediction models from MSA transformers. Studies in this area are currently being conducted.
Article Source: Paper Reference
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Riya Vishwakarma is a consulting content writing intern at CBIRT. Currently, she's pursuing a Master's in Biotechnology from Govt. VYT PG Autonomous College, Chhattisgarh. With a steep inclination towards research, she is techno-savvy with a sound interest in content writing and digital handling. She has dedicated three years as a writer and gained experience in literary writing as well as counting many such years ahead.





 Prediction.){kind=link}