

Saitama University, Japan, researchers developed a powerful method for observing the time-resolved behavior of biomolecules through high-speed atomic force microscopy (HS-AFM), thus improving the approach for estimating structural information from AFM data.
Deciphering dynamics of biomolecules
The surface geometry of a sample molecule is the only structural information present in HS-AFM pictures, though. It is crucial to infer latent three-dimensional structures from surface geometry in order to get a deeper understanding of a target biomolecule’s conformational dynamics. The rigid-body fitting to HS-AFM pictures is the foundation of the existing approaches for estimating the candidate structures. The present frame-by-frame rigid-body fitting analysis is expanded to many frames in order to make use of orientational correlations of a sample molecule between H frames in HS-AFM data caused by contact with the stage. The technique uses hidden Markov modeling to evaluate HS-AFM data, which are seen as time series data. As a test instance, the proposed technique demonstrates a more reliable prediction of molecular orientations than the frame-by-frame analysis using simulated HS-AFM pictures of the taste receptor type 1. The approach may be used to simulate conformational dynamics holistically utilizing HS-AFM data.
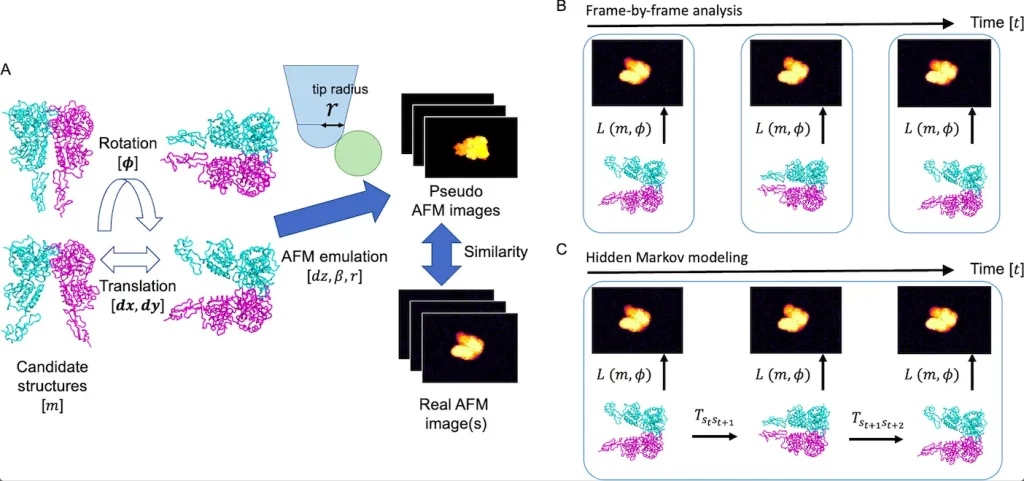
Image Source: https://doi.org/10.1371/journal.pcbi.1010384
In cells, biomolecules dynamically alter their structural configurations while carrying out a variety of vital life-supporting tasks. Using traditional experimental methods that focus primarily on static atomic structures, such as X-ray crystallography, it is challenging to discern such dynamic structural changes. Atomic force microscopy (AFM), a method that scans the surface of a sample entity with a sharp tip, is an alternate method. Recently, it has been feasible to see the dynamics of biomolecules in action by speeding up the AFM’s imaging rate. However, since a molecule’s surface geometry can only be determined with an AFM, it’s critical to derive three-dimensional structural information from the data.
HS-AFM for direct visualization of biomolecules
To better understand how biomolecules work, it is crucial to observe the conformational dynamics of the molecules in motion. Single-molecule measurements are effective methods among the various experimental measurement types because they can precisely characterize the heterogeneous fluctuations of biomolecules. Atomic force microscopy, optical tweezers, and Förster resonance energy transfer microscopies (FRET) are the measuring methods utilized in single-molecule measurements (AFM). AFM scans a molecule’s surface with a probe that has an acute tip to gather data on the surface morphology. Typically, the traditional AFM imaging rate is too slow to watch the conformational dynamics of biomolecules in action.
The high-speed AFM (HS-AFM) was created to get around this restriction by increasing the speed of scanners and the resonance frequency of cantilevers. We can directly study the behavior of biomolecules at high spatiotemporal resolution using HS-AFM. In fact, HS-AFM has been extensively used to observe the conformational dynamics of numerous biomolecules, including complex meta-structure protein interfaces of disordered regions, the conformational dynamics of CRISPR-Cas9 in action, myosin V walking, and the rotational catalysis of F1-ATPase.
Studying multi-frame images via hidden Markov modeling-based method
In this study, a frame-by-frame rigid-body fitting technique that uses hidden Markov modeling to evaluate numerous AFM pictures as time series data is shown to be a logical progression. Molecular states may be primarily described when modeling AFM data by a mix of structure and rotation. In order to capitalize on molecules’ propensity to interact with the substrate and maintain temporal correlation in the rotational direction, the modeling system has advocated constraining transitions in rotational space. The rotationally constrained Hidden Markov modeling, in contrast to the traditional frame-by-frame rigid-body fitting, can make estimates using the statistics of multi-frame images.
Limitations of the proposed method
The predicted accuracy using HS-AFM enhances in relation to the number of frames. As a result, genuine experimental AFM data might be modeled more accurately than with current techniques. Before the approach is used on real experimental data, there are a few problems that need to be fixed.
First, a rotational constraint threshold needs to be decided upon beforehand. Even after setting an arbitrary threshold for the quaternion distance in this investigation, it is difficult to predict in advance how much the rotation of a sample molecule varies per frame.
Another notable limitation is the absence of “correct” structures in the rigid-body fitting. In this case, it is crucial to review the possible structures. The flexible fitting strategy, a simple remedy, can search for the structures most matched to the pictures when dealing with sparse data, such as AFM images, but it is prone to overfitting. As was done in the analysis of CryoEM data, finding physically reasonable structures and avoiding overfitting may be accomplished in this situation by employing a Bayesian engine like MELD (CryoFold).
However, in order to produce structural ensembles that match AFM pictures, a range of ensemble refinements and techniques, such as the maximum entropy method and a Bayesian approach, would be helpful.
Extracting temporal heterogeneous information is fixed by single molecule measurements is a hurdle when using the ensemble refinements approach with single-molecule measurement data, such as HS-AFM. To effectively capture the heterogeneous information, time-series modeling based on the state space modeling examined in this study should be created in conjunction with the ensemble refinements approach.
The major emphasis of this work is on orientation calculations, however, it is crucial to utilize the tip shape that is closest to the accurate one in order to increase the accuracy of not just orientations but also structure estimations. Through the use of scanning or tunneling electron microscopy, the tip may be directly viewed (SEM and TEM). SEM and TEM, however, only offer 2D projections of a sample, making it challenging to regularly reconstitute the 3D shape of the tip using these methods. Furthermore, it’s possible that the tip’s shape and degree of damage will fluctuate during AFM studies. Therefore, using just AFM data, it is important to determine the tip shape at the moment of measurement.
Concluding remarks
Although the suggested approach was discussed in this study as a logical extension of rigid-body fitting, it might also be applied to integrative structural modeling. It is possible to remedy simulation mistakes brought on by model parameters by combining simulated and experimental data or to understand experimental data in new depth by utilizing simulation structures. Flexible fitting simulations have been utilized to successfully modify and understand experimental data in the examination of the CryoEM density map. To provide a thorough interpretation of the experimental results in the context of single-molecule measurements, Matsunaga and Sugita recently improved MSM, which was first built using simulation data. The integration of HS-AFM and simulation data might also be used to create a precise MSM. The potential dependency of structural dynamics on orientations is a challenge when building an MSM from AFM data. As was done in this work, it is only sometimes possible to merely lower the transition probability with regard to direction. To determine if interactions with the stage are the cause of outliers in the structural dynamics, it is required to examine the transition probability and MSM for each orientation.
Article Source: Paper Reference
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Riya Vishwakarma is a consulting content writing intern at CBIRT. Currently, she's pursuing a Master's in Biotechnology from Govt. VYT PG Autonomous College, Chhattisgarh. With a steep inclination towards research, she is techno-savvy with a sound interest in content writing and digital handling. She has dedicated three years as a writer and gained experience in literary writing as well as counting many such years ahead.





{kind=link}