
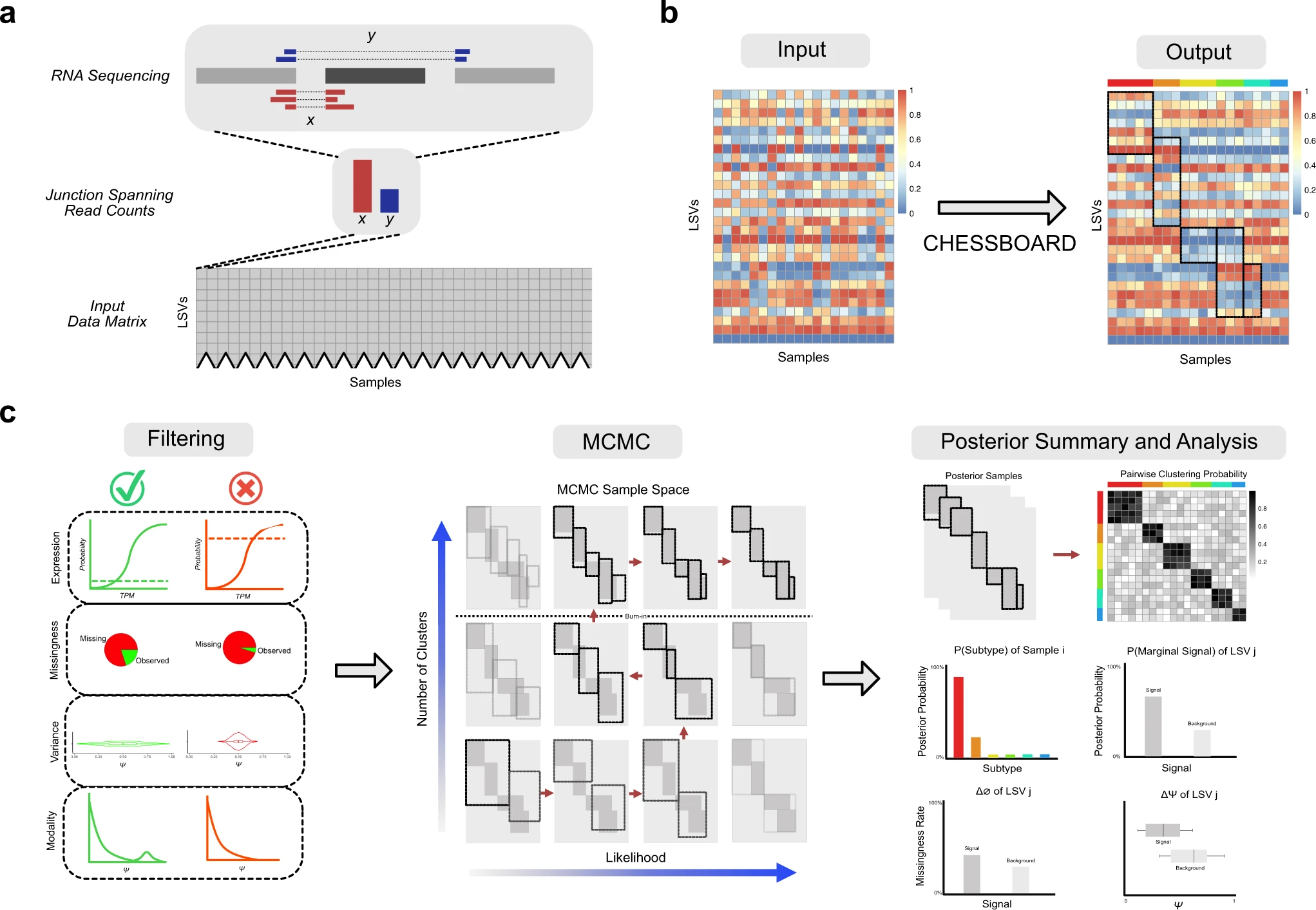
Finding cancer subtypes is a crucial first step in creating individualized care. In particular, RNA splicing change-based sub-typing has been encouraged by a number of recent studies. An unsupervised technique called CHESSBOARD was developed by scientists in the USA specifically for RNA splicing data. It identifies “tiles” in the data that are identified by a subset of distinctive splicing variations in a subset of patients. CHESSBOARD permits a variable number of tiles, takes into consideration the splicing quantification’s uncertainty, and has the ability to describe missing values as extra signals.
In order to evaluate the benefits of domain-specific modeling, CHESSBOARD is used for synthetic data. This is followed by a study of several leukemia datasets. This study demonstrates the consistency of the identified tiles across separate investigations, looks into potential regulatory factors, and assesses how they relate to well-known Acute Myeloid Leukemia (AML) mutations. In order to improve medication response correlation, mutation-based diagnostic tests are augmented with the identified splicing patterns, illustrating the potential therapeutic value of CHESSBOARD.
Present like a Pro! ExpertSlides – The secret weapon for your presentations!
RNA sequencing for revealing trancriptomic variations associated with complex disease.
It is frequently used to identify transcriptomic variations linked to complicated illnesses by analyzing RNA sequencing (RNA-seq) data from large patient populations. Such analyses can be conceptualized as either supervised or unsupervised learning tasks within the context of machine learning. In a supervised scenario, the goal is often to find transcriptome changes that serve as diagnostic indicators for a disease state or are highly linked with factors that have a bearing on patient outcomes. Finding latent substructures in the data that may be utilized to understand the genesis of diseases like cancer subtypes is a common task for unsupervised analysis. At the level of alternative splicing, one method of quantifying changes in the transcriptome is possible.
By using alternative splicing, various pre-mRNA segments can be eliminated while others are linked, or spliced, to create mature mRNA. Since hundreds of RNA binding proteins (RBPs) interact intricately to control alternative splicing, disruption from illness is a significant risk, particularly in malignancy. This study presents an unsupervised learning technique for finding substructures in a matrix of RNA splicing data from cancer patients because of the high correlation between splice variations and illness.
Driving factors that emphasize locating substructures in the RNA splicing cancer data.
First, the mutation burden is relatively low in malignancies like acute myeloid leukemia (AML), making it difficult to understand why important oncogenic pathways are disrupted by genetic alterations alone. According to a number of studies, the functionality of regulatory proteins associated with apoptosis and the control of cancer can instead be substantially perturbed by splicing abnormalities.
Second, it has been discovered that a lot of tumors are heterogeneous, with patients displaying a lot of variation in splicing measures. Recent studies have demonstrated that this variability might come from mutations that are rarely present in a significant portion of patients, even if part of this variability is likely caused by confounders such as batch effects.
Instead, they are seen in limited subgroups of individuals who have both trans-acting effects brought on by mutations in the splicing regulatory apparatus and cis-acting effects brought on by mutations at splice sites. These findings led to the development of a specific technique for locating “tile” substructures in the RNA splicing data matrix or patient subgroups with distinctive splicing variations in a subset of genes.
There is a growing demand for techniques for the unsupervised identification of splicing-based disease subtypes due to mounting evidence that splicing abnormalities are harmful in heterogeneous tumors like AML and B-ALL. CHESSBOARD is a technique that makes a number of contributions to the clustering and missing value modeling fields, which are both well-populated. Previous studies on tile discovery and biclustering techniques, in particular, were either not domain-specific or designed for different types of data modalities, such as gene expression and genetic mutations. Because of this, these algorithms fail to take into account important aspects of heterogeneous splicing cancer data, such as missing values and ambiguity in splicing quantifications. Additionally, the MNAR model developed by CHESSBOARD may be useful in areas well beyond RNA splicing or even clustering, such as techniques for dimensionality reduction like sparse probabilistic PCA or factor analysis.
In addition to the CHESSBOARD model, there are other techniques and tools to enable a more thorough analysis of the data. First, a pre-filtering and recursive clustering algorithm make it easier to analyze the full transcriptome. Recursive clustering was then utilized to identify alternative AML subgroup classifications that were highly linked with AML gene mutations. Secondly, putting an LSV ranking mechanism into place makes it possible to prioritize driver genes for use in later analyses like GSEA.
Although we used CHESSBOARD on a number of leukemia datasets, beatAML served as the main focus since it provided both a large number of samples and treatment response metrics. In beatAML, a single robust “signal” tile distinguished the dataset’s two primary patient subgroups and was highly repeatable in a different dataset. The disparity between tumor samples and ENCODE’s cell lines, the small number of RBPs ENCODE supports, and the intrinsic noise in the CLIP and KD tests are a few possible causes of this seemingly low proportion.
The mTORC1 pathway was found to be enriched in the genes-carrying events distinguishing the two BeatAML subgroups, and the potential functional ramifications of the subgroups were analyzed. The clusters may represent differences in cellular metabolism that might change drug susceptibilities in AML samples since mTOR is typically active in malignancy in a way that influences medication susceptibility.
Additionally, the signal tile represents a potentially less harmful subtype of AML. According to this theory, the background tile was characterized by SRSF1 misregulation, which has an impact on a number of oncogenes in the tile, including BIN1 and CASP9.
By identifying a different tile in the BeatAML data that corresponded with FLT3-ITD, NPM1, and CEBPA mutations—all of which are referred to together as normal karyotype AML— the value of CHESSBOARD’s recursive clustering was found in this study. The finding of this well-known subtype demonstrates the effectiveness of recursive clustering. By eliminating the dominating signal resulting from splicing variants brought on by the improper regulation of RBPs/SFs, the identification of a new tile structure linked to a distinct AML subtype with lower but still detectable splicing signals but a strong mutation signature was made possible.
Concluding Remarks
The first RNA splicing-specific method for signal recognition in heterogeneous RNA-seq datasets was created and is called CHESSBOARD. On numerous leukemia datasets, its usefulness was demonstrated by tying the found splicing tiles to putative regulators, medication responses, and well-known pathways. CHESSBOARD may be simply modified using a multiview model56 for alternative datatypes, including expression and the integration of multi-omics data. Furthering our knowledge of the function of splicing in complicated illnesses, it is also anticipated that the scientific community will make use of the open-source code and apply CHESSBOARD to a variety of additional analysis tasks in sizable, diverse cancer datasets.
Article Source: Reference Paper | CHESSBOARD python API
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Riya Vishwakarma is a consulting content writing intern at CBIRT. Currently, she's pursuing a Master's in Biotechnology from Govt. VYT PG Autonomous College, Chhattisgarh. With a steep inclination towards research, she is techno-savvy with a sound interest in content writing and digital handling. She has dedicated three years as a writer and gained experience in literary writing as well as counting many such years ahead.





{kind=link}