
Using read pairs with several SARS-CoV-2 variant-specific signature mutations as a reliable signal of low-frequency variants, scientists develop the bioinformatics technique known as COJAC (Co-Occurrence adjusted Analysis and Calling). The genomic sequencing, in conjunction with COJAC, can offer population-level estimates of the prevalence and fitness of emerging variants from wastewater samples earlier and on the basis of far fewer samples than from clinical samples. The proposed bioinformatics framework is applied routinely in large national projects in Switzerland and the UK.
Image Source: https://doi.org/10.1038/s41564-022-01185-x
The ongoing appearance of SARS-CoV-2 variants of interest and concern highlights the necessity of early detection and epidemiological surveillance of novel variants.
The scientists employed genome sequencing to track the population-level local distribution of the SARS-CoV-2 B.1.1.7 (Alpha), B.1.351 (Beta), and P.1 (Gamma) variants in 122 wastewater samples from three locations in Switzerland.
COJAC, a bioinformatics technique developed by scientists, leverages read pairs harboring several variant-specific hallmark mutations as a reliable signal of low-frequency variants.
A small outbreak of the Alpha strain in two Swiss cities was detected in wastewater up to 13 days before it was initially noted in clinical samples, according to the COJAC application.
After analyzing 1,339 more wastewater samples, the researchers were able to further demonstrate COJAC’s capacity to identify new variants for the Delta variant before they become widespread.
They demonstrate that replicate and closely-meshed longitudinal sequencing allows for robust estimation of not only the local occurrence but also of the transmission fitness advantage of any variant, whereas single wastewater samples sequencing data provide limited precision for the quantification of the relative prevalence of a variant.
They conclude that genome sequencing and their computational analysis can, sooner and with a lot fewer samples than clinical samples, provide population-level estimates of the prevalence and fitness of emerging variants from wastewater samples. In Switzerland and the UK, huge national initiatives frequently employ their framework.
Importance of Early Detection and Monitoring
The continued transmission and evolution of SARS-CoV-2 have led to several variants of interest and variants of concern (VOC), which may have varying degrees of impact on disease severity, diagnostics, treatment efficacy, and vaccination effectiveness.
Early detection and monitoring of the dissemination of local variants has therefore become a crucial public health task.
Variant Monitoring in Wastewater
Sewage collected at wastewater treatment plants (WWTPs) can contain viral RNA from SARS-CoV-2 infected individuals, and its quantity has been shown to correspond with case reports.
Additionally, wastewater samples can be used for genomic sequencing or reverse transcription quantitative real-time PCR (RT-qPCR) research to get a snapshot of the community’s circulating viral lineages and their diversity.
The prevalence of variants in wastewater has recently been demonstrated to correspond with clinical data. Because of this, genomic epidemiology based on individual patient samples may benefit from the effective and complementary strategy of variant monitoring in wastewater.
Complication of Detection
Although SARS-CoV-2 concentrations can be very low, samples may be enriched for PCR inhibitors, viral genomes are frequently fragmented, and sewage contains significant amounts of bacterial, human, and other viral DNA and RNA genomes, it is challenging to analyze wastewater samples for their SARS-CoV-2 genomic composition.
Amplification biases, sequencing mistakes, and missing phasing information also reduce the quality of the data gained from sequencing the mixture of viral genomes, making it more difficult to identify an emerging viral lineage that is only seen in a small percentage of infected people.
A New Framework
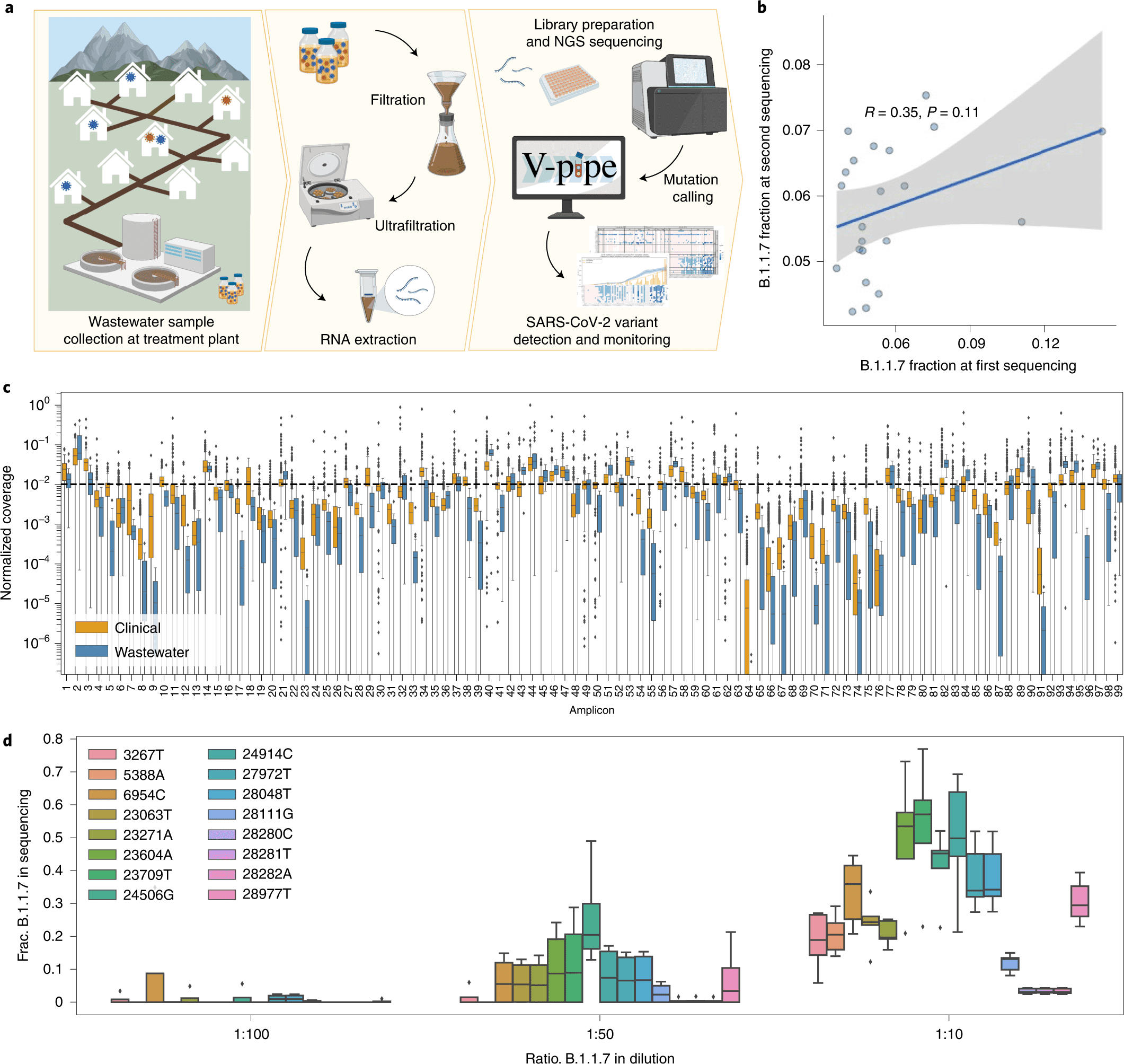
In this study, the scientists examined viral RNA isolated from raw influent samples acquired from several Swiss WWTPs using amplicon-based next-generation sequencing (NGS) data.
They carried out a number of repeat and spike-in tests to evaluate the reproducibility and quantifiability of sequencing data obtained from viral RNA isolated from wastewater.
Then, between December 2020 and mid-February 2021, they concentrated on a close-meshed time series in two major cities, as well as a ski resort during the holiday season (121 samples in total).
These samples span the time when Europe first encountered the Alpha, Beta, and Gamma variants. When the delta variant appeared, the researchers subsequently conducted validation analyses on 1,656 primarily daily samples from six WWTPs that were collected between January and September 2021.
A statistical technique for calculating the variant-specific transmission fitness benefit and quantitative variant monitoring (that is, the relative increase in the reproductive number of any genetic variant of SARS-CoV-2) of any SARS-CoV-2 variant was also developed by their team.
COJAC (Co-Occurrence adjusted Analysis and Calling) is a bioinformatics method for the early detection of low-frequency variants emerging in a population. On closely meshed time-series data, their methodology performs well.
The Endpoint
The scientists have illustrated the use of genomic sequencing of wastewater samples to identify, track, and assess SARS-CoV-2 genetic variants at the population level.
In particular, we have documented the local outbreak of the Alpha variant in two Swiss cities’ wastewater before it was found in clinical samples.
Despite very high clinical sequencing rates at the time in Switzerland, they increased their surveillance to six Swiss cities and discovered that in three of them, the earliest signal of the Delta variants in wastewater before the first local identification of the variant in clinical samples (between 66 percent and 94 percent of Swiss qPCR-positive samples were randomly selected for sequencing at that time).
In the cases where clinical samples gave the first local proof for the presence of the variant, the initial signal in wastewater emerged shortly after and at a time when the local prevalence of the Delta variant was still quite low.
The researchers have demonstrated the high correlation between the rate of clinical sequencing and the latency in variant discovery as compared to a wastewater-based analysis by subsampling the available clinical samples.
They have demonstrated that it is possible to discover early variants using wastewater samples, but we have also found that it can be difficult to interpret a single sample.
This is due to the difficulty in separating signal from noise in the sequencing data at first when only a tiny subset of hallmark mutations is often found at low frequency.
The technological difficulties encountered during the collecting and processing of the raw wastewater sample, the extraction of SARS-CoV-2 RNA, and its amplification are to blame for the high amount of noise in the data.
The findings imply that repeat sequencing and high sample density over time are essential components of enhancing the signal-to-noise ratio.
The scientists also created a method to strengthen the signal in each sample individually. As their existence in a sample represents a significantly stronger signal than individual mutations, This technique looks for concurrent signature modifications on the same read pair.
The Alpha variant, which features a number of very specific mutation pairs and even one triplet that may be found in this way, was particularly well-suited for identification by this method.
The first clinical evidence for the Alpha variant in Lausanne came 8 days earlier in the clinical samples that were retrospectively evaluated and 13 days earlier in the co-occurrence-based evidence.
Among the other variants they looked at, Beta and Gamma had a set of co-occurring signature mutations, and Gamma also had a set of mutations that were variant-specific.
Although the researchers found some early signs of these variants entering the Swiss population, neither of these two variants was able to displace the Alpha variant in Switzerland.
There are two sets of signature mutations for the Delta variant, one shared by B.1.617 and one unique to B.1.617.2, which the researchers employed for early co-occurrence-based detection.
In general, the exclusivity of co-occurrences and the presence of recurrent mutations in different lineages have a detrimental impact on the co-occurrence analysis’s utility for variant identification.
Shared signature mutations—by chance, convergence, or homology—are more likely to occur as variants proliferate and perhaps coexist in a population.
Deconvolution techniques will be helpful in this situation to separate the aggregate signals of co-occurring variants in wastewater.
Although the focus of this study was the introduction of known variants into a new population, the data they produced and the techniques we developed can, in theory, be applied to a de novo identification of circulating variants and the detection of cryptic variants in unsampled human or non-human animal populations.
The researchers have demonstrated that, in addition to early detection, sequencing data from wastewater samples can be used to track the regional prevalence of a variant, estimate its growth rate, and determine its fitness advantage for transmission earlier and with significantly fewer samples than when using clinical samples.
Additionally, wastewater samples have the advantage of representing cases in the data that are asymptomatic but undiagnosed and are routinely ignored in clinical sequencing.
Analysis and interpretation of sequencing data produced from wastewater continue to provide a number of difficulties.
As an illustration, the migration of individuals can make it difficult to relate the estimations from the wastewater to a particular local population. Like many other European nations, Switzerland has a high rate of commuting between its various areas and from its neighboring ones.
The high congruence between clinical and wastewater samples that they observe in terms of estimated prevalence and fitness advantage suggests that there may not be much difference between these two sources of information in terms of the general presence of infectious people who are frequently present in a city due to residence or daily commute.
The potential for different shedding profiles among variants, which may affect quantification from wastewater sequencing and thus may affect some of the inferred epidemiological properties of the variants, presents another difficulty.
In conclusion, this research has demonstrated how genomic analysis of SARS-CoV-2 variants in wastewater samples can support epidemiological research and supplement current methods based on clinical data.
The scientists have increased the scope of our ongoing sequencing work to include six wastewater treatment facilities around Switzerland, for which we provide real-time information to both the general public and neighborhood public health organizations.
Based on longitudinal sequencing of wastewater samples, their methods and ongoing sequencing effort offer a guide for quick, objective, and economic genomic surveillance of new SARS-CoV-2 variants.
Article Source: Jahn, K., Dreifuss, D., Topolsky, I. et al. Early detection and surveillance of SARS-CoV-2 genomic variants in wastewater using COJAC. Nat Microbiol (2022). DOI
Learn More About Bioinformatics:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}