
Scientists demonstrate that L452R and T478K mutations in the Delta variant interact synergistically with nearby residues to cause significant alterations in the spike/ACE2 interface, suggesting a single mode of action.

Understanding the molecular distinctions of emerging SARS-CoV-2 variants is essential because they raise questions about the capacity to endure the Covid-19 pandemic.
Using crystallographic structures and extended analysis of microsecond molecular dynamics simulations, the scientists investigated differences between the SARS-CoV-2 wild type and five variants that appeared in late 2020, focusing on the structure and dynamics of the spike protein interface with the human angiotensin-converting enzyme 2 (ACE2) receptor.
In contrast to WT and Epsilon, dihedral angle principal component analysis (PCA) revealed that the spike receptor binding domain (RBD) dynamics of the Alpha, Beta, Gamma, and Delta variants shared significant similarities.
Since the unique L452R and T478K mutations in the Delta variant are not in direct contact with the human ACE2 receptor, dynamical perturbation networks, and contact, PCA was used to identify the distinctive interface dynamics of the Delta variant.
The research findings demonstrate that the L452R and T478K mutations in the Delta variant interact synergistically with nearby residues to cause significant alterations in the spike/ACE2 interface; this suggests a single mode of action, which ultimately explains why it prevailed over earlier variants.
SARS-CoV-2 and the Variants Associated
By first infecting human pulmonary cells, the SARS-CoV-2 virus, linked to the Covid-19 pandemic, has spread throughout the entire planet.
The homotrimeric transmembrane spike glycoprotein (S protein, with 1273 residues in each monomer) and human angiotensin-converting enzyme 2 (ACE2) receptors interact specifically to carry out this crucial phase.
The receptor binding domain of the spike, which binds to the N-terminal helix of ACE2 with high affinity, is specifically responsible for this attachment to cells. This, in turn, enables conformational changes and membrane fusion between the viral and host cell membranes.
Due to its zoonotic origin, interspecies transmission, and human host adaptation, the creation of mutant strains (or variatiants), like in many other viral infectious illnesses, has ineluctably emerged.
Since the identification of the particular ACE2 receptor is the primary step in cell infection, mutations in the spike protein may provide higher or decreased infectivity potential, resulting in differences in transmission rates.
The characteristics of viral transmission, disease severity, and neutralization susceptibility have all been impaired by the swift global spread of variants of concern (VOC).
In the UK, the initial variant of concern was discovered in late December 2020. (Alpha variant, B.1.1.7 lineage). The independent emergence of a different variant (Beta, B.1.351) in South Africa was followed by the emergence of new variants in Brazil (Gamma, P.1), California (Epsilon, B.1.427/B.1.429), and lastly, India (Delta/Kappa, B.1.617.1/2/3).
In the summer of 2021, the hazards posed by the Alpha and Epsilon variant were reduced. The most recent variant of concerns (Omicron, B.1.1.529) was discovered in South Africa in November 2021 and quickly spread to other nations, where it is now the predominant type. The Delta variant had previously dominated for about a year before this.
Only predictions may be made from phylogenetic studies or binding free energy estimates because the processes by which these mutations modify the infectivity or severity of the disease are not well known.
Several mutations found in the spike RBD are frequently shared by most variants, such as N501Y and L452R, with the emphasis on the initial stage of viral infection or cell entrance.
On the other hand, some mutations are more recognizable, such as T478K, which was only found in Delta before the Omicron variant was discovered.
The recognition phase or the binding affinity between RBD and ACE2 receptors may be significantly altered by the physicochemical interactions between hydrophobic and charged residues.
For instance, it has already been demonstrated that the host ACE2 protein and S protein have less affinity when the mutation N501T is present in vitro.
Reports from the Investigation
In the study, the scientists present a thorough analysis of the atomistic interactions between the spike RBD domain and its human ACE2 receptor for the original SARS-CoV-2 virus and five SARS-CoV-2 variants that appeared in late 2020.
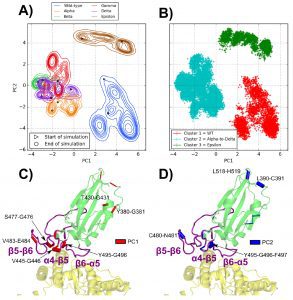
Image Source: https://doi.org/10.1021/acs.jcim.2c00350
In order to achieve this, the researchers concentrate on analyzing the main molecular interactions between spike and ACE2 using experimental structural data from the Protein Data Bank (PDB).
The contact changes between the wild type (WT) and the Alpha, Beta, and Gamma variants, located at 2.85, 2.63, and 2.80, respectively, are the first thing they look into.
Because the implicated structures are resolved at a lower atomic resolution that does not permit proper estimates of atomic contacts (i.e., >3 ), they did not compare those results with the available cryo-EM structures of the Delta and Epsilon variants.
Spike–ACE2 Association Triggers Initial Molecular Mechanisms
The development of vaccines, the design of RNA polymerase inhibitors, the investigation of the binding of small molecules of the RBD, the design of main protease inhibitors, and the clarification of the role of glycans in SARS-CoV-2 viral entry were all made possible by molecular dynamics (MD) simulations in the fight against the Covid-19 pandemic.
Notably, earlier research on the increased infectivity of variants examined the influence of mutations on antibody binding and found that the Beta variant’s mutations were allosterically communicating to one another.
The WT structure with the highest resolution at the time (PDB 6M0J) serves as the basis for modeling all variants in this study. From there, in silico mutations and structural equilibration are introduced.
Then, the scientists run MD simulations on several spike-ACE2 complexes in monomeric form (one unit of each protein). As a result, they examined the main molecular interactions between ACE2 and spike, paying particular attention to how various mutations affected the atomic contacts at the interface and the resulting binding dynamics.
Atomistic simulations are necessary because this information cannot be obtained directly from the crystallographic structural models present in the PDB (more than 200 X-ray or cryo-EM derived structures).
They used a number of methods, including dihedral angle principal component analysis (dPCA), static and dynamical perturbation contact networks (PCN and DPCN, respectively), and contact principal component analysis, to assess the MD trajectories and cross-compare them (cPCA).
The dPCA demonstrates that the distinct mutations in the Alpha, Beta, Gamma, and Delta variants cause identical rearrangements within the spike RBD that are incompletely duplicated in the Epsilon variant.
Even though these alterations are related to mutations (L452R and T478K), which affect residues outside of the interface, dynamical perturbation contact networks demonstrate that significant differences in the dynamics of the interface develop between the Alpha-Gamma group and the Delta variant.
Finally, the researchers demonstrate using cPCA how the combined impacts of the L452R and T478K mutations in Delta lead to a pattern of particular contact rearrangements that have a significant impact on the RBD/ACE2 interface.
A fundamental understanding of this important facet of viral infection is provided by this knowledge of the early molecular mechanisms activated by the spike-ACE2 connection, which may be highly helpful for the logical development of antiviral drugs.
The Endpoint
Based on the available crystallographic structures of the Alpha-Gamma variants of SARS-CoV-2, the scientists first analyzed the (static) networks of atomic contacts between the spike RBD protein and the ACE2 human receptor in this study, capturing the contact changes with respect to the WT and thus perturbations due to RBD mutations.
Microsecond MD simulations of the WT and Alpha-Epsilon variants have then been run in order to take into account the dynamical effects of RBD mutations on the contacts at the spike/ACE2 interface.
The primary similarities and differences between distinct spike RBD variants interacting with the human ACE2 receptor have been determined using a variety of MD trajectory analysis methods.
First, finding mobile RBD areas whose dynamics are changed by mutations was made possible by the analysis of protein essential movements based on backbone dihedral angles, or dPCA.
Backbone dihedral angles’ first main components correspond to motions in the α4−β5 loop, whereas their second principal components correspond to motions in the β5−β6 loop.
Three distinct tendencies have been noticed for the various MD simulations when taking into account these fundamental motions: The WT has a flexible α4−β5 loop and a tight 56 loop; for the Epsilon variant, the tightest β5−β6 loop was observed along with a partially flexible α4−β5 loop. The cluster involving the Alpha, Beta, Gamma, and Delta variants features a tight α4−β5 loop and a flexible β5−β6 loop.
It’s interesting to note that this grouping is related to the influence of the SARS-CoV-2 disease’s severity and transmissibility in the variants under study.
These findings imply that the effects of the L452R and N501Y mutations on RBD movements close to the interface are closely connected.
The dPCA of the Epsilon variant reveals an interdependence between these mutations, as these motions cannot be adequately replicated in the absence of the T478K mutation.
This modification in the RBD’s flexibility close to the interface may be the first step toward making it easier for the spike trimer to bind to many ACE2 receptors. However, the dPCA analysis was unable to separate the dominant Delta variant from the others in 2021.
Then, by examining the dynamical perturbation contact network and concentrating on the RBD/ACE2 interface, the researchers were able to partially restore the specificity of the Delta variant.
The Alpha-Gamma variants that share the N501(Y) mutation, which promotes specific perturbations for the interface contacts of Y501 with K353 and Y41 residues, while the rest of the interface contacts remain largely preserved, showed many similarities between the WT atomic contact network and those of the Alpha-Epsilon variants.
However, despite the absence of interface mutations, significant contact alterations at the interface have been seen in the Delta variant. The T478K and L452R mutations must have indirect (but significant) impacts on the interface for there to be any interface contact alterations in Delta.
The propagation of contact disturbances caused by the T478K and L452R mutations in the Delta variant was eventually clarified by the subsequent cPCA analysis.
This investigation demonstrated how the T478K mutation affects the contacts of a hydrophobic cluster (consisting of residues Q474, T478, I472, V483, and F490) located around the C480-C488 disulfide bridge inside the RBD’s β5−β6 loop and encourages the development of a G485-C488 backbone hydrogen bond.
The locations of residues F486 and N487 are altered as a result of this rearrangement, increasing their interface contacts with ACE2’s two helices.
In addition, the presence of residue R452 and the altered F490 contacts caused by the T478K mutation are both affected by the L452R mutation. In the wild-type (WT), residues F490, L492, and L452 are involved in another hydrophobic cluster.
At the interface of Delta with residue K31, which was only hydrogen bound to residue Q493 in the WT, residues F490 and L492, in turn, form a triple hydrogen bond. Residue F490 is crucial to the transmission of contact modifications caused by the simultaneous T478K and L452R mutations that result in cooperation in producing the interface perturbations reported in Delta because it is a member of both the aforementioned T478- and L452-related hydrophobic clusters.
The scientists’ findings draw attention to the Delta variant’s unique method of action, which may ultimately explain why it prevailed over earlier variants.
A synergistic long-range effect of several mutations, such as that reported here for the Delta variant, must also be determined for the recently discovered Omicron variant because it carries the same T478K mutation alongside the E484A mutation.
Article Source: Gheeraert, A., Vuillon, L., Chaloin, L., Moncorgé, O., Very, T., Perez, S., … Maigret, B. (2022). Singular Interface Dynamics of the SARS-CoV-2 Delta Variant Explained with Contact Perturbation Analysis. Journal of Chemical Information and Modeling, 62(12), 3107–3122. DOI: https://doi.org/10.1021/acs.jcim.2c00350
Learn More About Bioinformatics:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}