
The prediction of drug-target interactions (DTI) plays a significant role in drug development by helping in the identification of new drug targets, the repurposing of current medications for new indications, and the prediction of potential adverse drug effects. This article discusses a research study that presents a novel computational model for predicting DTIs. The model consists of three phases: feature extraction, feature selection, and classification. The suggested model predicts DTI with a high degree of accuracy, making it a cost- and time-effective alternative to experimental approaches in drug development.
Different approaches used to predict DTIs
Predicting drug-target interactions (DTIs) is essential for drug development since drugs usually interact with at least one protein in the body, and misconceptions about these interactions can lead to negative consequences. Experimental methods for finding DTIs are expensive and time-consuming, hence computational alternatives to speed the process have been proposed. There are three kinds of computational techniques, each with advantages and disadvantages: ligand-based, docking-based, and chemogenomic-based. Feature-based, kernel-based, and similarity-based machine learning algorithms can also be utilized for DTI prediction. These methods aim to give useful insights into drug mechanisms and speed the discovery and development of novel medications.
Kernel-based approaches are good at modeling nonlinear interactions but lack interpretability and efficiency for modeling huge datasets. Feature-based techniques extract physical, chemical, and molecular features in order to disclose inherent features for enhanced interpretability. There are two types of feature-based approaches: deep learning and classical. Deep learning techniques employ the protein sequence and drug structure to extract features in several layers and predict DTIs.
Previous works on the prediction of DTI
Jiajie Peng and colleagues employed the learning representation graph to provide a framework for DTI prediction in recent research, whereas Anna Cichonska et al. suggested an approach with multiple pairwise kernels for efficient memory and time learning. Similarity-based approaches and kernel-based methods are also prevalent in this discipline. Yet, there are still issues with machine learning-based approaches, including difficulty choosing negative samples, overly straightforward experimental conditions, and inadequate descriptive features. These limitations make it difficult, from a pharmacological standpoint, to differentiate possible medication mechanisms from their functions.
Feature-based approaches
Much research has been conducted on feature-based approaches in the field of predicting DTIs. These approaches are novel in four areas: feature extraction, feature selection, balance, and the development of novel classifiers. Utilizing fingerprints for the electro-topological status of drugs and APAAC of target proteins, combining drug and protein features to provide per drug-protein pair features, and applying new computational models such as PSHOG gradient and PSSM matrix for feature extraction have all been explored by researchers. In addition, balancing techniques such as SMOTE have been proposed to work with imbalanced data. Moreover, new classifiers have been created utilizing protein sequence data and the GIST feature. These feature-based techniques have assisted in addressing data inconsistencies, data class imbalances, and the extraction of essential drug-target properties during the prediction process.
Several approaches have been developed for predicting drug-target interactions (DTIs), including the use of a Rotation Forest classifier and LBP feature extraction, a novel computer model, and a Fuzzy classifier. Unfortunately, the feature extraction procedure has not received enough attention despite its usefulness in boosting discrimination and verification rates. The high dimensionality of features and data imbalance are further issues that must be addressed. Deep learning-based DTI models require a vast amount of data and have a heavy computational burden, thus, traditional approaches that extract features from drug and protein sequences have been examined.
Steps involved in the proposed method
The suggested technique comprises many steps, including the extraction of features from proteins and drugs, the selection of features, and the training of a model.
In the first stage, distinct features are derived from protein sequences and the structure of medications. A molecular fingerprint for a drug is retrieved from its structure, which is a Boolean substructure vector. The protein sequence features include EAAC, EGAAC, DDE, TF-IDF, k-gram, BINA, PSSM, NUM, PsePSSM, and PseAAC. In the second stage, the features of each drug-protein combination are integrated, and a “one” or “zero” label is assigned based on whether or not they interact. In the third stage, irrelevant and noisy features are eliminated, and the number of input variables is reduced using the IWSSR approach. Lastly, the rotation forest model is trained to identify DTIs using the specified features. A flowchart demonstrates the approach.
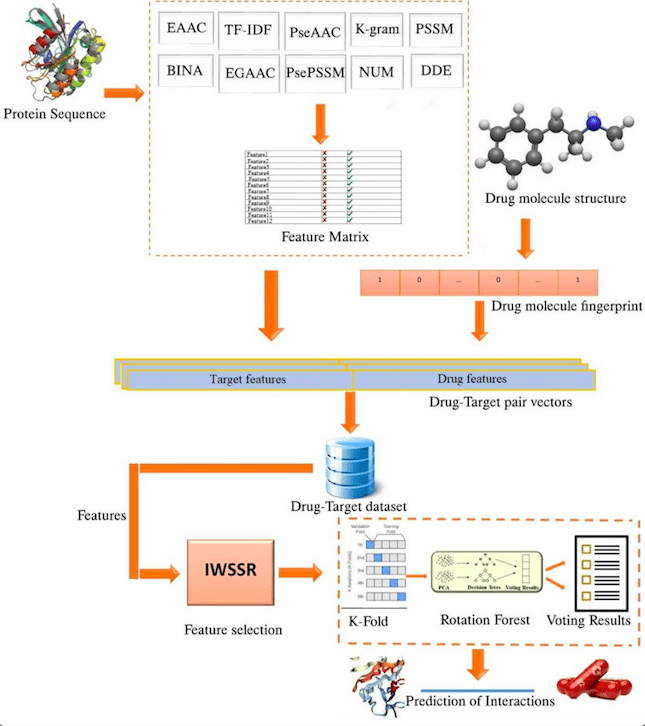
Image Source: https://doi.org/10.1038/s41598-023-30026-y
Analyzing the results
The study analyzed the efficiency of a suggested approach using four assessment criteria: accuracy, sensitivity, precision, and Matthew correlation coefficient. The Gold Standard dataset used in the study was obtained from http://web.kuicr.kyoto-u.ac.jp/supp/yoshi/drugtarget/ and was divided into four datasets: enzymes, ion channels, G-protein-coupled receptors, and nuclear receptors. The study employed ten feature-extraction techniques to extract various protein features, which were subsequently analyzed using the rotating forest model.
The PsePSSM feature was shown to have the highest distinguishing power and detection rate, whereas PSSM, PseAAC, and BINA features all showed good performance. The combination of varied features enhanced the classification model’s ability to recognize DTIs. Three of the many ways of integrating features had superior performance. The study employed ROC diagrams to summarise the results, and the space under the curve (AUC) was calculated to confirm the capability of making predictions.
Comparison with other methods
The proposed method for protein sequence analysis was assessed against other available methods utilizing the same dataset. According to the findings, the suggested technique attained the greatest levels of precision, sensitivity, specificity, and MCC. One of the reasons why it worked better was because it defined and picked more exact features, which led to more precise findings. The method also evaluated balance in classes and did not indicate a bias towards the majority class. In contrast, one of the other techniques demonstrated a statistically significant difference in its specificity and sensitivity values. Therefore, the proposed technique performed better than alternative methods for analyzing protein sequences.
Conclusion
The research paper discussed here presents a DTI (drug-target interaction) prediction model based on protein features employing wrapper feature selection. The model consists of three phases: feature extraction, feature selection, and classification. Protein sequence and drug fingerprint information are used to extract several features, including EAAC and PSSM. The combined features are then processed via the IWSSR wrapper feature selection technique to identify the most pertinent features. Lastly, DTIs are predicted using a Rotation Forest classifier. The suggested model has demonstrated satisfactory results and is comparable with other techniques in the literature.
Article Source: Reference Paper
Learn More:




Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





, based on protein features, using wrapper feature selection.){kind=link}