
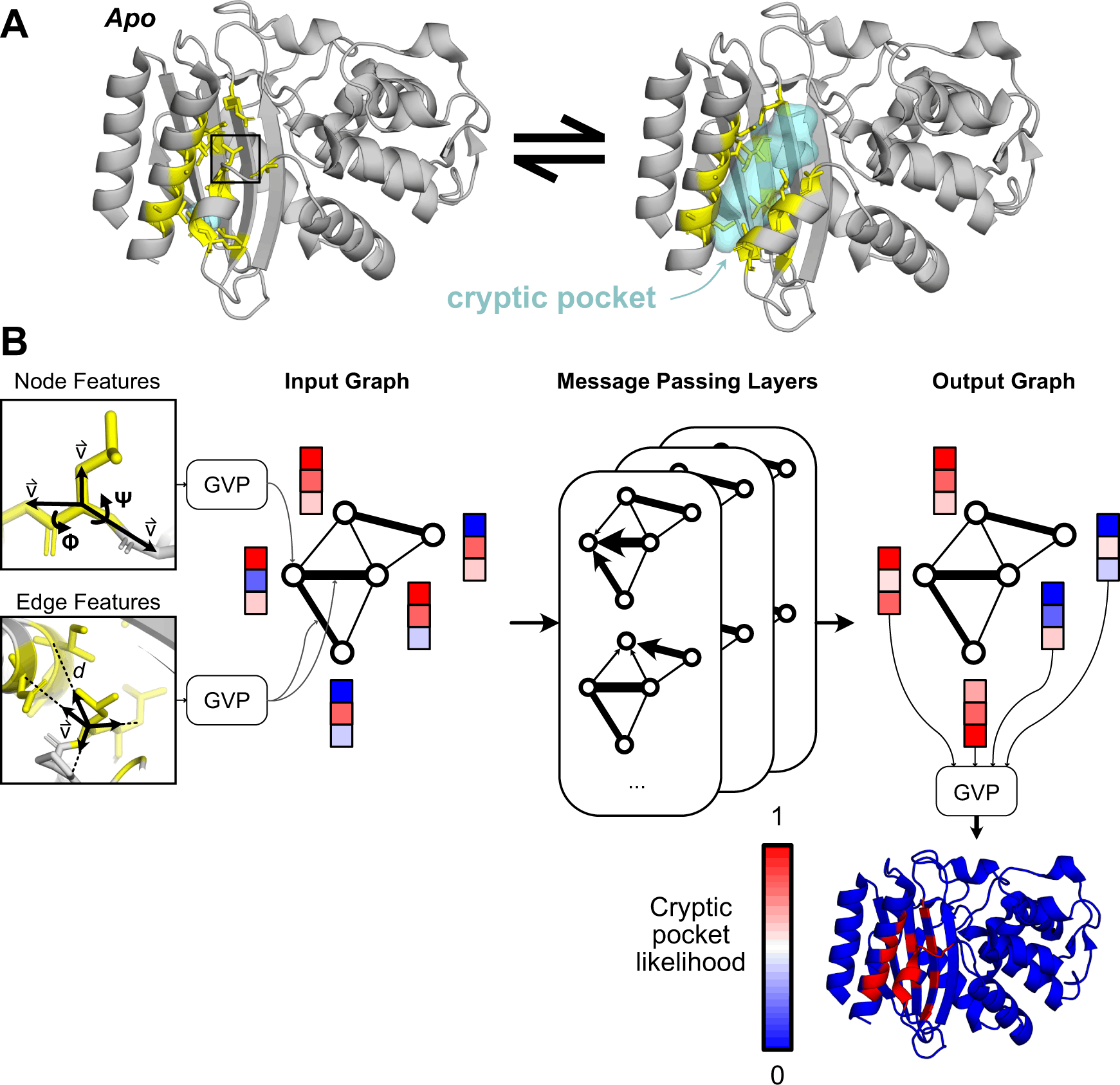
The discovery of cryptic pockets has widened the scope of drug development by permitting the targeting of proteins previously deemed undruggable due to the absence of pockets in their ground-state structures. However, detecting cryptic pockets takes a significant amount of time and effort. PocketMiner is a machine learning tool built by researchers from the University of Pennsylvania. It is a trained graph neural network that predicts where pockets will likely appear during molecular dynamics simulations. Over fifty percent of proteins considered to lack pockets based on published structures likely have cryptic pockets, drastically broadening the druggable proteome.
Cryptic Pockets and Need for PocketMiner
Cryptic pockets are locations on proteins that are not evident from the protein’s original structure but can emerge as a result of structural fluctuations. These cryptic pockets can be targeted for drug development and offer various advantages over orthosteric site targeting. Nonetheless, locating and targeting cryptic pockets is difficult and frequently requires accidental discovery or computationally costly molecular dynamics simulations.
The researchers propose using simulations to determine whether each residue in a protein structure can reorient itself to participate in a cryptic pocket as a result of thermal variations. The proposed method of training does not require instances of ligands bound in cryptic pockets; rather, it employs structural ensembles derived from molecular simulations to get thousands of cryptic pocket opening events for training.
PocketMiner is a trained graph neural network that predicts where pockets are likely to form in molecular dynamics simulations using simulations from multiple studies. On a dataset of experimentally developed structures containing known cryptic pockets, the accuracy of PocketMiner was evaluated, and it was able to recognize various forms of cryptic pockets. Finally, PocketMiner was applied to the complete human proteome in order to identify new cryptic pockets that could serve as prospective therapeutic targets.
Using Molecular Dynamics Simulations
The scientists reviewed the effectiveness of molecular dynamics simulations in capturing the creation of known cryptic pockets in proteins. Sixteen proteins known to develop cryptic pockets from apo (ligand-free) starting structures were subjected to adaptive sampling MD simulations. The simulations demonstrated that the majority of cryptic pockets emerge in close proximity to known ligand-binding sites in just ten parallel simulations lasting 40 nanoseconds each. The findings show that a minimal quantity of simulation data may be sufficient to detect cryptic pockets, and those machine learning models trained to predict cryptic pocket formation across short simulation time periods may be able to identify cryptic sites in ligand-free experimental structures.
The authors hypothesized that sufficiently extended simulations would sample comparable pocket opening events, and they validated their intuition by analyzing the consistency of pocket openings across independent simulations. Using labels generated for each residue in each 40 ns window’ starting structure based on whether that residue participates in a cryptic pocket at any point in the next 40 ns of simulation, they trained a graph neural network and a 3D convolutional neural network. The model with the best performance was the GVP-GNN, with an average test PR-AUC of 0.44 ± 0.12. Given the native, folded state of a protein, it may be able to identify places where cryptic pockets occur without computing intermediary states (e.g., via MD simulations).
Development of PocketMiner
To determine if their models could detect cryptic pockets, the researchers compiled a dataset of cryptic pockets. They filtered through the Protein Data Bank to identify 38 apo-holo protein structure combinations containing 39 cryptic pockets that had substantial root mean square deviations between apo and holo. This resulted in the PocketMiner dataset, a collection of cryptic pockets created by several sorts of conformational alterations. The dataset contains a wide variety of structural rearrangements and serves as a difficult standard for evaluating machine learning techniques.
Using ligand-free crystal structures, PocketMiner can accurately predict the locations of cryptic pockets (hidden binding sites) on proteins. The model was trained with several labeling techniques and assessed with both positive and negative instances of cryptic pocket creation. It outperforms comparable algorithms in terms of predicting fewer false positives and having a faster prediction time across multiple classes of cryptic pocket openings.
Using PocketMiner on more than 10,000 human genes, the authors identified several thousand proteins with predicted cryptic pockets. They proved the efficacy of PocketMiner by detecting cryptic pockets in proteins involved in the Jak/Stat system, which has been linked to a number of malignancies, including PIM2 and WNT2. Simulations of these proteins revealed the existence of cryptic pockets and proposed possible therapeutic targets.
Conclusion
Scientists have developed PocketMiner, a graph neural network that predicts the locations of cryptic pockets in folded protein structures. Unlike previous methods that required molecular simulations as input features, PocketMiner was trained using simulation data that incorporated pocket opening events. This resulted in a model with better performance and speed, making the algorithm for recognizing cryptic pockets faster and more accurate. In addition, the researchers discovered that the majority of known ligand-binding cryptic pockets might be identified with just 400 ns of aggregate unbiased simulation without producing a large number of false positives. This method can facilitate structure-based drug creation and should be accessible to a broad range of scientists.
Article Source: Reference Paper
Learn More:




Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





{kind=link}