
Due to the absence of adequate theoretical models and the enormous protein sequence space, the data-driven design of proteins with particular functions is difficult. The Protein Transformer Variational AutoEncoder (ProT-VAE) is a novel deep generative model for protein engineering that combines the capabilities of variational autoencoders and transformers to provide an accurate, quick, and transferrable model. The ProT-VAE model was implemented using NVIDIA’s BioNeMo framework and has been verified by retrospective functional prediction and prospective design of new protein sequences. The model can provide a framework for machine learning-guided directed evolution campaigns for the data-driven creation of new synthetic proteins.
Advances in Deep Learning Networks for Protein Design
Synthetic biology has long been in search of synthetic proteins with engineered functionality. The huge size of protein sequence space and the absence of adequate theoretical models for the sequence-function link present substantial challenges. Deep-learning networks such as variational autoencoders, generative adversarial networks, and transformers are data-driven and empirical methods that have been developed to solve this challenge. These models excel at functional prediction and have the potential to create new proteins with non-natural functionalities.
Protein design models need accurate sequence-function learning, generative sequence design, quick and transportable model training, and unsupervised and semi-supervised training. Traditional models were constrained by handcrafted features and simple linear models. Modern deep-learning networks like VAEs and transformer-based models can learn all amino acid associations and are generative. Convolutional or recurrent layers can feature protein families and minimize latent space dimensionality, although learning long-range mutational connections may be difficult.
The Protein Transformer Variational AutoEncoder is a deep learning model that combines the advantages of transformers and variational autoencoders (VAEs) for efficient and precise protein creation. The transformer component enables learning of long-range correlations and variable-length sequence data processing, while the VAE component offers a low-dimensional latent space suited for interpretation, annotation, and guided generative design. The ProT-VAE model is created and trained using the NVIDIA BioNeMo framework, and it has promising results in both retrospective computational prediction tasks and future protein design.
Integrating Transformers and Variational Autoencoders
There are several examples of deep neural network designs that combine transformers and variational autoencoders to address a variety of application areas, including narrative completion, music analysis and production, text modeling, layout identification, and protein engineering. The ProT-VAE model is intended for protein engineering activities and has been evaluated using synthetic protein sequences in wet lab experiments.
The ProT-VAE model consists of a transformer-based encoder that produces a high-dimensional encoding of a protein sequence, which is subsequently compressed using a VAE. The latent space embedding is then supplied to an attention-based decoder stack, which produces a new protein sequence with the appropriate properties. While the encoder and decoder may be transferred across jobs, only the VAE component of this model must be retrained for different protein engineering activities. In addition, the model may be trained unsupervised or semi-supervised, which is advantageous when experimental measurements are unavailable for all protein sequences.
The ProT-VAE model highlights the possibility of integrating transformers and VAEs for protein engineering tasks, and its performance in wet lab tests has been confirmed.
Training the ProT-VAE model
The ProT-VAE model is a variational autoencoder that generates new proteins with the desired properties by directly working on the non-aligned sequences of a homologous protein family. It contains three essential elements:
- A pretrained transformer-based T5 encoder and decoder model.
- A generic dimensionality reduction block that compresses the transformer’s hidden state into a more parsimonious intermediate-level representation.
- A family-specific maximum mean discrepancy variational autoencoder that compresses the intermediate-level representation into a low-dimensional latent space.
The model is initialized and trained from scratch for each target protein family of interest for a given design assignment. The resulting low-dimensional generative manifold enables more efficient production of latent space with fewer overall network parameters.
Using the MSE aim, scientists produced a pretrained ProtT5 model and pretrained intermediate dimensionality reduction layers on Uniprot. Using an Adam optimizer with a learning rate of 0.0001, family-specific variational autoencoder layers were trained on unaligned sequences of a single homologous protein family, with 15% of locations randomly masked and 20% of masks corrupted to distinct amino acids. The goal was to reassemble the original sequence.
Accurate Prediction and Alignment-free Designing
The Src homology 3 (SH3) protein family and the phenylalanine hydroxylase (PAH) enzyme were used to evaluate the ProT-VAE model. The SH3 family is involved in diverse cellular signaling functions, and the ProT-VAE model was able to learn an interpretable latent space organizing protein sequences by ancestry and function, make accurate predictions of protein function, and generatively design novel synthetic sequences with function comparable to or superior to that of natural sequences. The model accomplished this without the necessity for repeated sequence alignment. This highlights the ProT-VAE model’s potential for data-driven functional protein design.
Using a protein dataset, the ProT-VAE model was optimized to predict protein function without requiring a multiple sequence alignment (MSA). The capacity of the model to predict functional performance and recreate phylogenetic separation within the latent space was examined. The outcomes demonstrated that the ProT-VAE model could reliably predict protein function and distinguish phylogeny among paralog groups, indicating its potential for alignment-free generative design and manufacturing new synthetic proteins. The performance of the model was evaluated in the context of creating a therapeutic protein.
Substrate Specificity and Predictive Capacity
Using a dataset of 20,000 sequences from the aromatic amino acid hydroxylases (AAAH) family, especially phenylalanine hydroxylase, the ProT-VAE model was tested (PAH). For assessment, sequences were projected onto a 6D latent space after the model was fine-tuned. The model was able to successfully arrange substrate specificity annotations and distinguish and cluster the most annotated functional substrates. The generative aspect of the model was evaluated by reconstructing sequences and determining the percentage of identity between the encoded sequence and the original sequence. Based on the learned functional structure of the latent space, the model was also able to predict substrate specificity with high precision.
Using the same latent space, the predictive capacity of the ProT-VAE model is assessed on several tasks, such as the decrease of immunogenic reactions. The AAAH dataset is subdivided at the phylum level by phylogeny, and the embedding is annotated by phylogeny. High reconstruction accuracy of all classes and high prediction accuracy of phylum labels using the k-nearest neighbors classifier are exhibited by the model. These findings are encouraging for the de novo generation of proteins with specialized functionality for a certain host, such as the humanization of therapeutic proteins with specific activity or specificity.
Designing of Proteins
By fitting a Gaussian around hPAH in the latent space and enforcing a maximum similarity to any natural sequence in the training data, a conservative approach was chosen to generate sequences. The selection of 190 sequences for experimental synthesis and testing. The figure depicts the activities of the proposed sequences as measured by a plate-based assay. ProT-VAE is capable of MSA-free de novo design of synthetic PAH proteins from a generative and interpretable latent space and creating unique, highly active, and varied sequences.
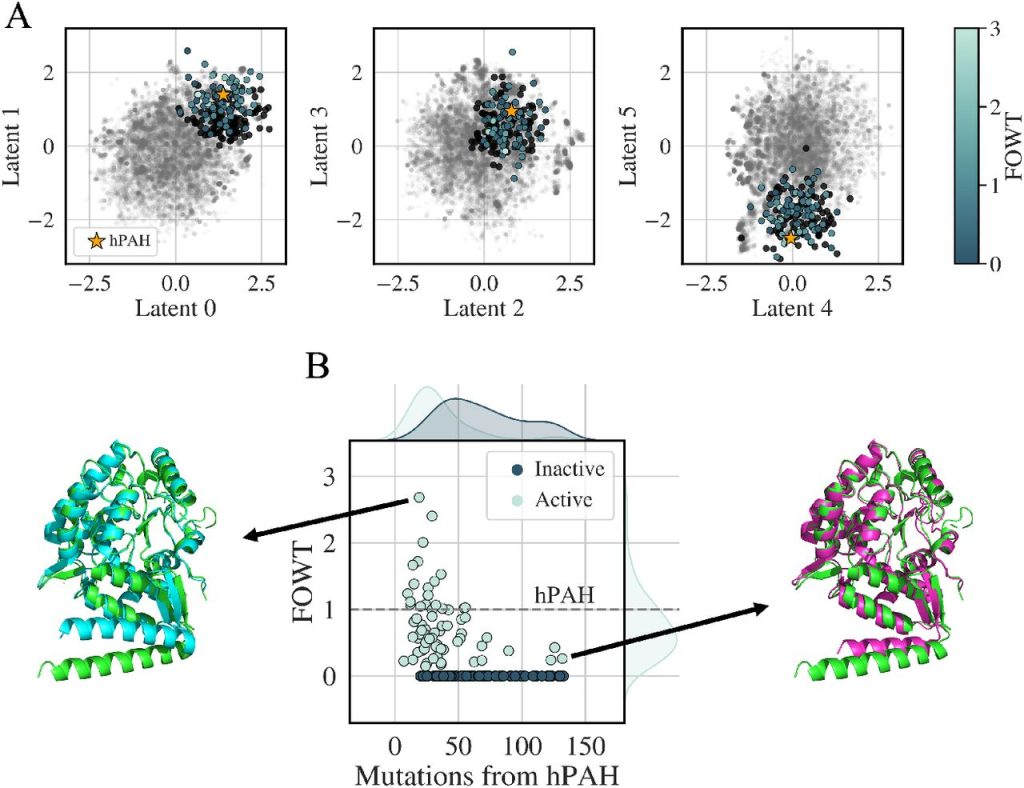
Image Source: https://doi.org/10.1101/2023.01.23.525232
Conclusion
ProT-VAE is a novel model that combines the favorable characteristics of transformers and VAEs to represent the sequence-function link in protein engineering. ProT-VAE is accurate, generative, and capable of quick and transferable model training in an unsupervised or semi-supervised manner. It employs a pre-trained transformer encoder and decoder stack and generic compression/decompression blocks to map between a high-dimensional latent space and a compressed representation that is fed to a task-specific VAE. The VAE is retrained for every protein engineering activity and generates a low-dimensional latent space that improves the comprehension and generative design of functional synthetic sequences. The model is portable and may be used to produce any protein from unaligned sequences.
Article Source: Reference Paper | Reference Article
Important Note: BioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





{kind=link}