
Cancer therapies often become ineffective due to the development of resistance in the tumor cells against the treatment. One potential strategy to overcome this resistance was treating the patients with a combination of drugs. Scientists conducted a study to improve cancer treatment by developing deep-learning models for drug synergy prediction. The models were designed to identify effective drug combinations for patients by considering the synergistic effects of two or more drugs. The study’s results will help lead the development of new deep-learning models for drug synergy prediction in the future.
Combating Drug Resistance
The phenomenon of drug resistance in cancer treatment was a major obstacle to reaching successful outcomes. Tumor cells can acquire resistance to anti-cancer treatments owing to a variety of circumstances, including pre-existing features or an adaptive response to therapy. One of the main drivers of drug resistance was intra-tumoral heterogeneity, which led to the emergence of subpopulations of cells with different characteristics and varying sensitivity to drugs.
Using numerous drugs instead of just one can assist in overcoming drug resistance. Drug combinations can avoid pre-existing resistance mechanisms more readily and prevent the development of acquired resistance mechanisms. Furthermore, certain combinations of drugs may have synergistic effects, which means that the combination of drugs produces greater effects than would be expected based on the individual effects of each drug. This can improve therapeutic efficacy without increasing drug dosage, potentially avoiding increased toxicity.
Quantifying Drug Synergy
Drug synergy can occur through a variety of mechanisms, including the drugs having the same target, multiple targets belonging to the same biological process or pathway, or multiple targets belonging to related pathways. Synergy can also occur when one drug’s activity boosts another drug’s transport, permeation, distribution, or metabolism.
However, it was difficult to quantify drug synergy. There are a variety of drug combination reference models with varying underlying assumptions, which can result in contradictory outcomes. The dose-response profiles of individual compounds and variations in the experimental design can also have an impact on the determination of combination effects. To evaluate the efficacy of drug combinations and their potential synergistic effects in cancer treatment, it was necessary to carefully consider and interpret the results of the experiments.
To find effective anti-cancer drug combinations, high-throughput cell viability assays can be used to screen a large number of drug combinations at various concentrations. However, it was impractical and costly to screen all possible drug combinations. Based on screening experiments and other relevant data, computational methods like biological network analysis-based approaches and machine learning (ML) models can be used to prioritize drug combinations and predict drug synergy. Using publicly available drug combination screening data and omics datasets describing the screened cancer cell lines, a number of ML models have been developed. The amount of work required for experiments to find drug combinations that work against cancer can be greatly reduced with these resources.
Deep-learning Models and Neural Networks for Drug Synergy Prediction
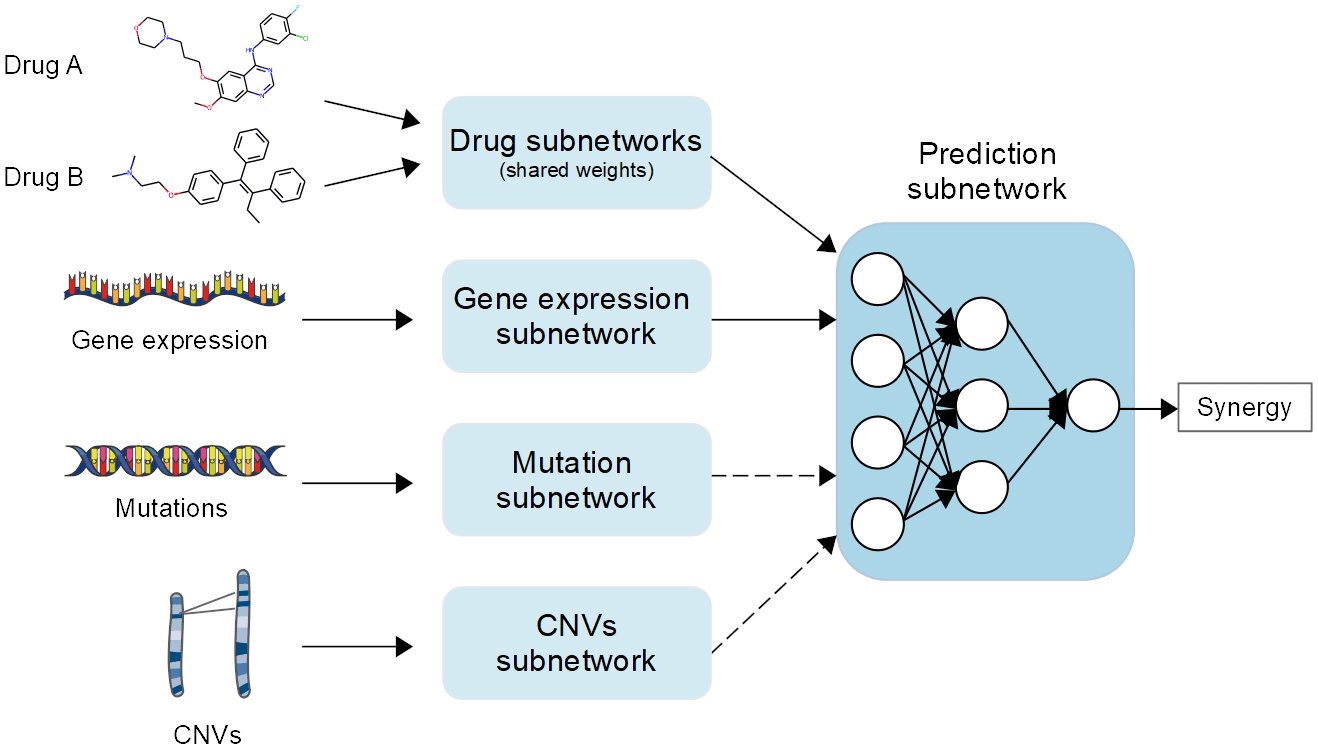
DL models can handle large amounts of high-dimensional and noisy data without requiring extensive feature selection, making them particularly useful for learning complex, non-linear functions. Because of this, they are excellent candidates for the creation of drug synergy prediction models, which aim to predict the effectiveness of drug combinations in treating diseases. In recent years, a number of drug synergy prediction models based on DL have been proposed. For instance, Preuer et al. presented DeepSynergy, a feedforward, fully connected deep neural network that predicts drug synergy by utilizing data on gene expression and chemical features. Xia and co. developed a multimodal DL model to predict the NCI ALMANAC project’s cell lines’ growth inhibition.
Additionally, efforts have been made to turn drug descriptors into images, making it possible to model drug synergy with convolutional neural networks (CNNs) rather than fully connected networks. Researchers in the field developed this approach, which was known as the REpresentation of Features as Images with NEighborhood Dependencies (REFINED). Graph neural networks (GNNs) trained on graphs containing information on interactions between drugs in a combination, between drugs and their targets, or between genes or proteins in cell lines have also been used by some researchers.
Feature reduction
Drug synergy prediction models normally utilize various sorts of information, for example, drug features and extra cell line data like hereditary or proteomics information. However, adding more features can make the model more complicated, so it’s important to figure out which input data types are better at predicting drug synergy.
Feature reduction methods like PCA and autoencoders are frequently used to reduce the dimensionality of omics data from cell lines. Predefined gene lists give you more control over the selection process and may make it easier to interpret the models from a biological perspective. However, model generalization may be limited by restricting gene features to known drug targets in the training set. Information-driven approaches can conquer this issue and distinguish the most useful highlights from the information. The specific objectives and constraints of the study should guide your choice of feature reduction technique.
Comparison with baseline models
The study’s authors created a number of models to predict drug synergy and compared their results to a random baseline model that always predicted the training set’s average ComboScore value. They also built baseline models to figure out how important it was to include certain types of input data in predicting drug synergy, like the chemical characteristics of drugs and omics features of cell lines.
The three baseline models were:
- cell lineone hot + drugsone hot: trained exclusively with one-hot encoded cell line and drug identifiers
- cell lineone hot + drugsECFP4: used one-hot encoded cell line identifiers and extended connectivity fingerprint (ECFP)4 fingerprints as input features, removing cell line omics features
- exprDGI + drugsone hot: used one-hot encoded drug identifiers and genes with known or potential interactions with drugs screened in the ALMANAC project (drug-gene interaction (DGI) genes) as features, removing drug features
The authors discovered that the random baseline model performed worse than any of their models. They also found that the cell lineone hot + drugsECFP4 model, which used one-hot encoded cell line names instead of omics features, had lower scores than some models trained on actual gene expression data.
Gene expression subnetworks
The study compared different types of gene expression subnetworks and found that fully connected subnetworks trained on log-transformed and min-max scaled FPKM values using predefined gene lists produced the best performance scores. Due to its superior scoring metrics and potential for improved interpretability, the exprDGI gene selection method, which came in second place in performance, was selected for the subsequent tests. Performance scores were lower when dimensionality reduction techniques like WGCNA or UMAP were used.
Mutations and Copy Number Variations
When mutation and CNV data were included, the model’s performance was slightly reduced, indicating that the model was less accurate at predicting disease-gene associations.
Interestingly, the model trained with gene-level mutation data outperformed the model trained with pathway-level mutation features slightly. This might be on the grounds that summarizing the mutation information at the pathway level considers a bigger number of genes and gives a more complete image of genetic modifications inside a pathway.
When using gene-level mutation data, the model may miss relevant genetic information, as it only considers a subset of genes.
Selection of important features
The SHapley Additive ExPlanations (SHAP) interpretability framework was used by the researchers to identify the most crucial features for predicting drug response with DL models. The main 20 features for the DL model were all drug features, showing that drug features are the most important for drug response prediction in the ALMANAC dataset. The atom environments that characterize every one of the ECFP4 bits that show up among the main 20 most significant features are likewise analyzed. The SHAP framework can be used to provide explanations for specific examples, and a specific example involving the SR cell line, Topotecan hydrochloride, and Gefitinib was analyzed. The top 20 gene expression features are also shown.
Conclusion
The study found that cell line features in the ALMANAC dataset were less accurate predictors of drug combination effects than drug features. The models mostly used gene expression data to tell different cell lines apart but didn’t use it to find specific synergy biomarkers. The performance of various compound representation methods was comparable, but combining various drug representations may improve model performance.
Other cancer-specific gene lists and pathway propagation methods could further enhance the predictive capacity of the models, and prior biological knowledge was useful for feature selection. However, selecting genes with drug targets or DGIs may limit the model’s generalizability.
Article Source: Reference Paper
Learn More:




Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





{kind=link}