

Scientists at Carnegie Mellon University, USA, introduced PeptideBERT, a language model that is capable of predicting the properties of proteins and peptides solely based on sequences provided to it without requiring structural data. In this study, activities related to hemolysis, non-fouling, and solubility of peptides are taken into consideration for predicting the proteins. The model is trained to analyze shorter sequences using transformers, and it displays state-of-the-art (SOTA) performance when predicting hemolytic and non-fouling properties.
Significance of peptides and their properties
Peptides are ordered sequences of amino acids with a varying number of residues. The arrangement of the residues and the length of the peptide chain plays a significant role in determining the properties of proteins and peptides. The sequence also determines the nature of interactions between the peptide and the environment surrounding it. This model has been extensively trained on the hemolytic, solubility, and non-fouling properties of peptides because they are therapeutically important. The order of the amino acids in peptides also affects these characteristics.
- Hemolysis: It refers to the breaking down of red blood cells (RBCs), i.e., their lysis. The properties of peptides that can potentially induce lysis in RBCs are studied for clinical purposes. It is important to understand the influence of the sequence arrangement on such properties to develop safer and more efficient treatment methods against bacterial infections. Peptides with hemolytic properties can regulate cell proliferation, modulate immune responses, and induce cell death as well.
- Non-fouling: It is the ability of peptides to resist interactions that are not specific to them. Fouling, described as the accumulation of particles on the surface of the membranes of cells, generates resistance to interacting with the external environment. Studying this property in peptides helps formulate more effective drug delivery methods, produce more durable biomaterials, and engineer medical devices with higher precision.
- Solubility: It refers to the capability of the peptide to dissolve in a given solvent. It is important to measure and study how this property affects the potency of drug delivery systems.
All three properties mentioned above contribute to advancing biomolecular engineering research and drug development; therefore, it is vital to produce quicker and more accurate methods using modern deep-learning approaches for greater productivity.
Previously used methods: Quantitative Structure-Activity Relationship (QSAR) Models
They are used to correlate structural properties with the sequences of peptides. They predict the properties of several classes of peptides, such as antioxidant peptides, inhibitory peptides, and antimicrobial peptides. Three QSAR-based models have been popularly used for carrying out research in this domain:
- DSResol: It is used for predicting properties related to the solubility of peptides. It exceeded the accuracy rates exhibited by ProteinSol, DeepSol, and SoluProt, all of them being computational and deep learning approaches, with an accuracy of 75.1%. It uses dilated convolutional neural networks (CNNs) to analyze the k-mers of amino acids. The length of a segment of peptide sequences is denoted by ‘k,’ and these segments are referred to as k-mers. The disadvantage of this method is that it needs the protein structure to perform predictions, and it is not easy to access the structure for every peptide to be analyzed. Thus, the number of peptides that can be used in this method remains limited.
- MahLooL: It exhibits accuracy rates comparable to those of DSResol and even outperforms it for sequences of very short lengths, with an accuracy of 91.3%. It extensively correlates sequences using bidirectional Long Short Term Memory (LSTM) networks, a variation of recurrent neural networks (RNNs) that can learn long-term dependencies.
- HAPPENN: It is a SOTA model for performing hemolytic property predictions for peptides. It has an accuracy of 85.7% and uses features that are selected through a collection of Random Forests combined with Support Vector Machines (SVMs).
PeptideBERT: Transformer-Based Language Model as an Alternative
Peptide sequences can be considered to be words, and a collection of them can make up a ‘language’ of amino acids. These ‘words’ translate into proteins with meaningful functions and properties. Using this track of thought, PeptideBERT, a large language model (LLM), has been trained to use attention mechanisms for quickly forming connections between segments of sequence data in a textual format. Two key components involved here are transformers, which contain attention mechanisms, and a pretrained model, ProtBERT. PeptideBERT has been trained on a large dataset of protein sequences to fine-tune the entire model. Repeated patterns are learned through sequences present in the datasets, which help predict proteins with greater rates of accuracy than previously used models. The dataset only considers sequences that consist of L- and canonical amino acids.
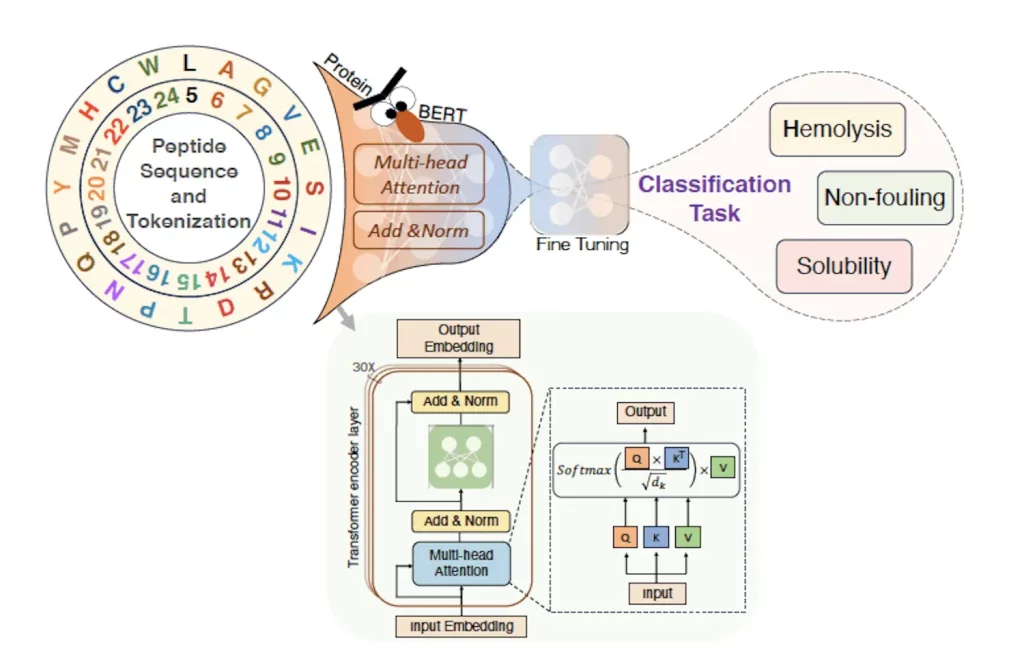
Image Source: https://arxiv.org/abs/2309.03099
Figuring out which peptides cause hemolysis and which ones do not is the hardest part of predicting their hemolytic properties. This is because the effects are seen on charged parts of cell membranes. To solve this problem, an approach that compiles databases that contain information on antimicrobial peptides (AMPs) is taken, referred to as ‘data augmentation.’ AMP recognition plays a vital role in discerning which peptides lead to hemolysis and which don’t, enabling their safe use in treating bacterial infections. They are selective, specific, and have negligible toxicity, offering several medical advantages. The peptides are categorized based on their hemolytic properties. This method increased accuracy exponentially, giving SOTA predictions for hemolytic as well as non-fouling properties. Peptide sequences of short length were used to study non-fouling properties.
Lower accuracy rates—around 70%—were recorded while assessing solubility properties because the analyzed dataset largely consisted of insoluble peptides. There is a possibility that accuracy can be increased by incorporating a significant number of soluble peptides into the dataset.
Conclusion
PeptideBERT has exhibited state-of-the-art predictions when analyzing the hemolytic and non-fouling properties of peptides, outperforming other methods in two of the three properties examined in this study. It exhibited competitive performance in predicting solubility as well. It needs to be noted that the dataset used here was confined to clinical and antimicrobial applications, and whether the model performs similarly in other domains is yet to be seen. The predictions given by this model show potential use in peptide research and applications, as well as in diverse domains ranging from biotechnology to drug discovery.
The necessary code for PeptideBERT can be accessed at GitHub
Article Source: Reference Article
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.





{kind=link}
[…] PeptideBERT: A Transformer-based Language Model for Predicting Peptide Properties using Amino Acid S… […]