
High-Throughput Sequencing (HTS) has drastically transformed the world of genomics by allowing rapid and accurate detection of genomic variations at the level of individual base pairs. However, the presence of technical artifacts (hidden non-random error patterns affecting the accuracy of variant identification) degrades HTS data quality. Scientists from the National Institute of Biomedical Genomics, India, have developed Mapinsights, a comprehensive toolkit to tackle the problem of artifact identification. Mapinsights conducts quality control (QC) analysis of sequence alignment files and demonstrates success in detecting outliers based on artifacts of HTS data with higher precision than the existing methods. The scientists tested Mapinsights on standard open-source datasets and observed that the toolkit effortlessly identified a variety of issues, such as errors pertaining to sequencing cycles, sequencing chemistry, sequencing libraries, and even sequencing depth (number of times a specific DNA region is sequenced).
A Glimpse into the Background of HTS
HTS has been gaining applicability in a wide variety of fields apart from biomedical sciences, such as agriculture, microbiology, human evolution, medicine, and many more. But, because of the humongous amount of data it generates, it has a high overall error burden. If left unidentified, these errors can become exceedingly costly, as they can become a part of decision-making processes in fields that impact human lives.
In the context of HTS data, the main types of errors that can possibly arise are:
- Pre-sequencing Errors: These errors occur during library preparation (converting nucleic acids into a sequencing platform-compatible format). One example is transversion due to ultrasonic shearing.
- Sequencing Errors: arise during the sequencing process itself. A few examples include overlapping and optical imperfections.
- Data processing Errors: These errors arise during data analysis due to the limitations of bioinformatics tools or the reference genome’s incompleteness.
- Systematic Errors: These errors include non-uniform depth of coverage (DCOV) or error profiles associated with specific sequencing platforms.
Some bioinformatics tools have features that can identify technical errors and detect outliers, but they are not devoid of shortcomings. Tools such as Qualimap2, Picard, Samstoolstats, FastQC, and Alfred fail to focus on base mismatches and are unsuitable for multisample analysis. Further, tools such as SeqControl and CovReport do not take into account variability and other imperative aspects regarding gene depth, particularly at the exon level.
Mapinsights: The Future of HTS Data Analysis is Here
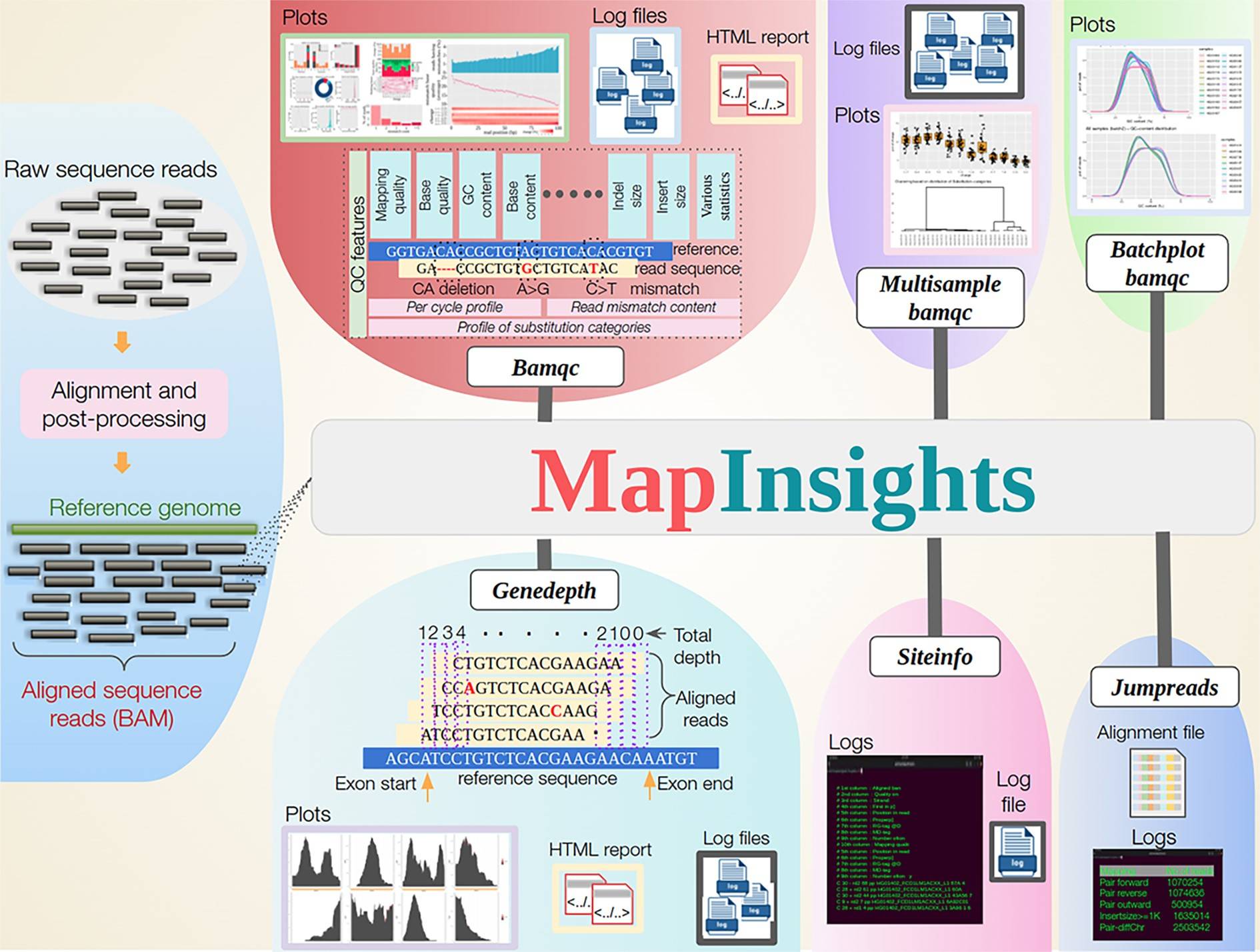
Researcher Analabha Basu and his team have developed a toolkit called Mapinsights. Mapinsights offers a range of modules designed to analyze and evaluate HTS data quality at different levels.
- The “BAMQC” module performs a comprehensive QC analysis of alignment files, providing both common and novel QC features. It allows users to assess crucial metrics, such as mapping quality, substitution patterns, and base quality across the alignment.
- The toolkit goes beyond single-sample analysis by introducing modules like “MULTISAMPLE-BAMQC” and “BATCHPLOT-BAMQC,” enabling comparison analysis across multiple samples. This functionality helps evaluate consistency among samples and detect outlier data points, providing valuable insights into the quality and reproducibility of the sequenced data.
- One notable feature of Mapinsights is the “GENEDEPTH” module, which estimates the exon-wise depth of coverage for genes. This information is critical for variant calling (identification of differences between the reference genome and sequenced sample), as low coverage regions may lead to missed mutations. By identifying such regions, Mapinsights aids in optimizing sequencing experiments and improving the capture efficiency of design probes.
- Mapinsights also includes the “SITEINFO” module, which queries the alignment file to obtain site-specific information. This feature enables researchers to investigate specific genomic positions that help detect true variants and reject false ones.
- And lastly, the “JUMPREADS” module validates structural variants and extracts suitable reads for downstream analysis.
Mapinsights: One Tool, Many Features
One of the key strengths of Mapinsights is its ability to identify sequencing artifacts, errors, and biases across different sequencing platforms. By analyzing open-source data, the toolkit reveals important insights into the performance and limitations of various sequencing technologies.
For instance, the analysis using Mapinsights identifies outlier samples in the 1000G-phase3 datasets with an inflated rate of G > T substitutions compared to C > A substitutions. These observations suggest the presence of sequencing artifacts, specifically oxoG errors, which occur due to heat, shearing, and metal contaminants.
Mapinsights also highlights the relationship between per-cycle substitution rate and mismatch base quality. As the sequencing cycle progresses, the end of the reads tends to exhibit more mismatches. This phenomenon is attributed to phasing and pre-phasing problems in Illumina platforms, inadequate flushing of non-incorporated nucleotides, and issues related to adapter content and erroneous end repair.
The toolkit further identifies specific errors and biases in recently launched sequencing platforms, such as MGI and AVITI. These platforms exhibit unique error patterns, including A > G and T > C mismatches in MGI data and reads with higher mismatches in AVITI data. The underlying mechanisms behind these errors and biases require further research, and Mapinsights serves as a valuable tool for investigating and understanding such new sequencing artifacts.
Mapinsights also offers valuable features that aid in variant calling and data interpretation. By differentiating reads that overlap with benchmarked variant sites from those that overlap with non-benchmarked sites, the toolkit provides insights into mapping quality, substitution categories, per-cycle profiles (representation of the distribution of reads across the genome at each sequencing cycle), and read mismatch content. Logistic regression analysis demonstrates that Mapinsights features can effectively separate these two classes with high accuracy.
While Mapinsights provides a comprehensive assessment of data quality, the integration of sophisticated deep learning approaches, such as DeepTrio and DeNovoCNN, could further enhance accuracy in variant classification. These deep learning tools are trained with known sequencing errors, technical artifacts, and mapping errors and can improve the identification of true variants versus sequencing artifacts.
Mapinsights also aids in understanding data variability by portraying the variability in the depth of coverage across different exons of a gene. Insufficient depth of coverage may lead to missed mutations, and Mapinsights’ gene-wise coverage estimation helps researchers identify potential regions of interest that require further investigation.
Conclusion
Mapinsights is an innovative and comprehensive QC toolkit that addresses the challenges associated with evaluating HTS data quality. By providing a systematic and in-depth analysis of sequencing artifacts, errors, and biases, Mapinsights empowers researchers to optimize their experimental design, improve variant calling accuracy, and gain a deeper understanding of data variability.
The toolkit’s modular architecture, open-source nature, and integration into HTS analytical pipelines make it a valuable asset for researchers in the genomics community. With its ability to analyze data from various sequencing platforms, including recently launched technologies, Mapinsights enables researchers to identify platform-specific error patterns and contribute to the advancement of sequencing methods and techniques.
As genomics continues to evolve rapidly, tools like Mapinsights will play a crucial role in ensuring the reliability and accuracy of HTS data, ultimately driving discoveries and advancements in the field of genomics.
Article Source: Reference Paper | Mapinsights source code is publicly available on GitHub
Learn More:




Neegar is a consulting scientific content writing intern at CBIRT. She's a final-year student pursuing a B.Tech in Biotechnology at Odisha University of Technology and Research. Neegar's enthusiasm is sparked by the dynamic and interdisciplinary aspects of bioinformatics. She possesses a remarkable ability to elucidate intricate concepts using accessible language. Consequently, she aspires to amalgamate her proficiency in bioinformatics with her passion for writing, aiming to convey pioneering breakthroughs and innovations in the field of bioinformatics in a comprehensible manner to a wide audience.





{kind=link}