
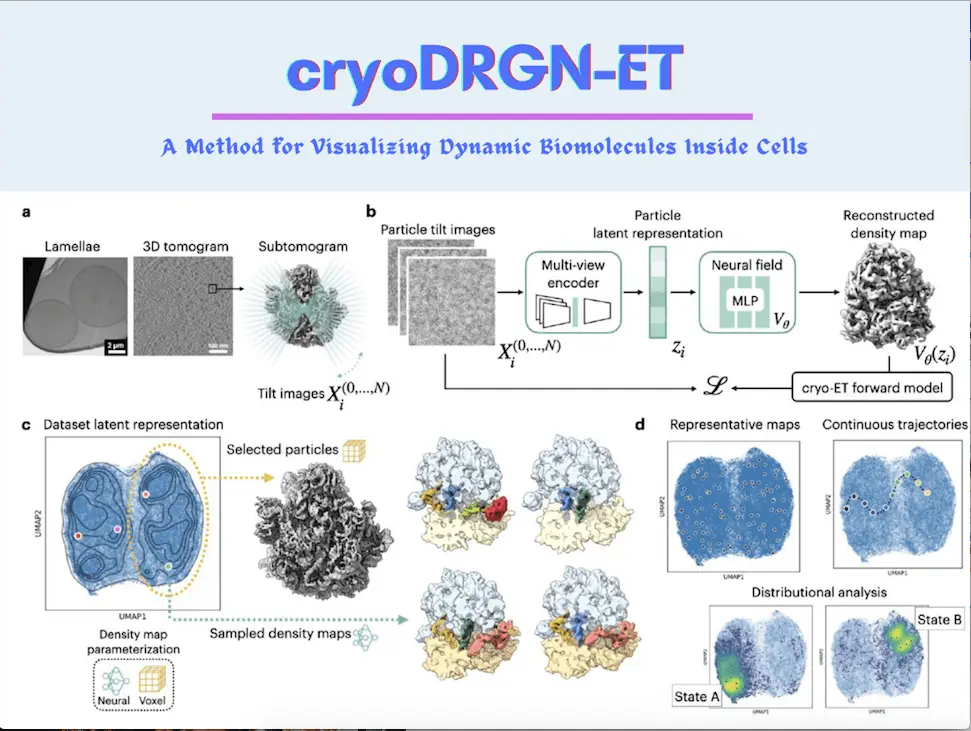
The advancement of cryo-electron tomography (cryo-ET) has opened novel avenues for visualizing the intricate structures of dynamic macromolecular assemblies within their natural cellular surroundings. Despite its potential, analyzing structural diversity within sub-tomogram data poses a challenge. A groundbreaking machine learning strategy, cryoDRGN-ET, has emerged to decipher the structural intricacies and kinetics of biomolecular complexes within cryo-ET sub tomograms, thanks to the collaborative efforts of researchers from Princeton University, Columbia University, Google (USA), Max Planck Institute (Germany), and Thermo Fisher Scientific (The Netherlands). By directly learning from tilt series images of sub-tomograms, this method employs deep generative models to reconstruct 3D density maps, representing a wide array of compositional and conformational states. The efficacy of cryoDRGN-ET is exemplified in its application to unveil the real-time translational dynamics of prokaryotic ribosomes. Notably, this approach unravels the spectrum of functional states adopted by S. cerevisiae ribosomes during translation elongation within cellular contexts.
Maximizing the Potential of cryo-ET with cryoDRGN-ET
Cryo-electron tomography (cryo-ET) is an advanced imaging method that offers a comprehensive view of structures ranging from cellular to molecular scales. By digitally combining a series of tilted images of intact cells or thin sections, cryo-ET achieves three-dimensional visualization of whole cells with nanometer precision. Computational post-processing of the resultant 3D tomograms, coupled with segmentation and subtomogram reconstruction algorithms, further enhances resolution to sub-nanometer levels. This facilitates the detailed examination of macromolecular structures and their contextual placement within native environments.
Despite its potential, analyzing structural diversity within subtomogram data poses a challenge. Cryo-ET-specific imaging traits, like low signal-to-noise ratios in individual tilt images and inherent variations in biomolecular complex conformation and composition, necessitate specialized subtomogram reconstruction methods. Present workflows, often reliant on 3D classification, necessitate manual steps, substantial computational resources, and assumptions about the number of states present.
In contrast, cryoDRGN-ET leverages a deep generative model directly trained on 3D density maps from particle tilt images. Comparable to the single-particle approach (cryoDRGN), cryoDRGN-ET’s model accommodates various sources of heterogeneity – compositional shifts, continuous conformational dynamics, and imaging induced artifacts. Validated on bacterial ribosome data, cryoDRGN-ET agrees quantitatively with previous analyses while unveiling continuous motions and membrane-linked conformations in a unified model. The researchers also employ cryoDRGN-ET to uncover the native landscape of the S. cerevisiae eukaryotic ribosome. This powerful technique is accessible as part of the cryoDRGN software package version 3.0, fostering broader applications in structural biology.
How cryoDRGN-ET Performs Its Magic
The generative neural network technique for analyzing heterogeneous cryo-electron tomography (cryo-ET) subtomogram data is introduced by CryoDRGN-ET. The method employs a standard cryo-EM image formation model specially adapted for tomography; it considers multiple tilt images from various angles of a single particle. CryoDRGN-ET amalgamates these tilted images using a multiview encoder to construct an implicit embedding zi that represents each particle’s conformational state. The generative model produces corresponding 3D density maps, which can be projected into 2D using estimated parameters. Using a maximum likelihood objective, it compares the generated projections with observed tilt images and handles factors such as signal attenuation in the contrast transfer function (CTF) due to electron dose.
This method adapts to large subtomogram datasets, enabling heterogeneity analysis via techniques like principal component analysis (PCA), UMAP visualization, and conformational interpretation after training. Latent embeddings generate representative density maps, which assist in exploring conformational diversity. This method also streamlines the systematic investigation of compositional and conformational variations by identifying particle classes that represent distinct states; it also allows trajectory mapping through latent space sampling as well. Homogeneous reconstruction with voxel-based backprojection can verify the validity of observed states.
Evaluating the Features of cryoDRGN-ET
The cryoDRGN-ET approach was evaluated using an in situ dataset of M. pneumoniae bacterial ribosomes treated with chloramphenicol (Cm), and its performance was compared to conventional 3D classification methods. Multiple tilted images of ribosome particles were processed through a multiview encoder to create a latent embedding representing each particle’s conformational state, employing a cryo-EM image formation model extended for tomography. The generative model then generated 3D density maps, which were compared with observed tilt images using a maximum likelihood approach.
The method’s efficiency was tested with an 18,466-particle dataset, excluding outliers and poor-quality images. The analysis showcased the technique’s capability to reproduce known compositional and conformational variations in bacterial ribosomes, capturing distinct translational states and their features, such as tRNA occupancy, elongation factors, subunit rotations, and local dynamics. Remarkably, cryoDRGN-ET produced major translational states in agreement with previous studies and quantified the relative occupancy of each state. Validations were performed through conventional homogeneous reconstruction and class assignments.
The method also revealed additional protein factor variability and larger-scale background variations, including ribosomes associated with cell membranes and polysomes. Importantly, cryoDRGN-ET achieved these results without multiple rounds of 3D classification or user-provided masks. This single-round approach presents a significant advancement over previous methods, demonstrating its potential for efficiently characterizing heterogeneous structural states in cryo-ET subtomogram data.
In a comprehensive demonstration, the cryoDRGN-ET technique was applied to a substantial cryo-electron tomography (cryo-ET) dataset of S. cerevisiae eukaryotic ribosomes obtained from cryo-plasma focused ion beam milling (cryo-PFIB). This analysis successfully uncovered the native structural landscape of the ribosome, showcasing its ability to identify known translational states, factor-binding events, continuous conformational changes, and spatial background variations. Through the method’s selective use of tilt images for enhanced resolution, the training process was efficiently executed on the extensive dataset of 119,031 particles.
CryoDRGN-ET revealed the separation of particles into distinct classes, including rotated and non-rotated small subunit (SSU) states, as validated through homogeneous reconstruction. Subsequent training runs specialized in delineating translational states, exposing evidence of functional states and their relative populations within the ribosomes. These states were validated by reconstructing and fitting atomic models, confirming expected tRNA and factor density distributions.
Additionally, cryoDRGN-ET uncovered broader compositional heterogeneity, membrane-bound ribosomes, polysomes, initiation factors, stalk proteins, and fungal-specific elongation factors, all of which were confirmed through further reconstructions. This analytical approach not only enabled efficient heterogeneity analysis of ribosomal states but also provided insights into various structural complexities within the context of native cellular environments, presenting a novel framework for in situ structural exploration.
Conclusion
cryoDRGN-ET has emerged as a transformative tool in the realm of cryo-electron tomography, offering a paradigm shift in the analysis of sub-tomogram heterogeneity. Through the potency of deep neural networks, this method unveils intricate density maps characterized by compositional and conformational dynamics, providing unprecedented insights into cellular macromolecular machinery. Its successful application to both prokaryotic and eukaryotic ribosomes showcases its versatility and reliability in capturing translational states and structural complexities within native environments. Remarkably, cryoDRGN-ET’s unbiased per-particle heterogeneity estimation presents a pathway to dissecting intricate relationships between conformational states, binding factors, and spatial context. As it redefines the landscape of structural analysis, its integration with advancements in ab initio reconstruction offers a promising avenue to unlock the full potential of visualizing dynamic cellular processes at the molecular level.
Article Source: Reference Paper | cryoDRGN Software is available on GitHub in version 3.0.0-beta.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Neegar is a consulting scientific content writing intern at CBIRT. She's a final-year student pursuing a B.Tech in Biotechnology at Odisha University of Technology and Research. Neegar's enthusiasm is sparked by the dynamic and interdisciplinary aspects of bioinformatics. She possesses a remarkable ability to elucidate intricate concepts using accessible language. Consequently, she aspires to amalgamate her proficiency in bioinformatics with her passion for writing, aiming to convey pioneering breakthroughs and innovations in the field of bioinformatics in a comprehensible manner to a wide audience.





{kind=link}